
An Alternative Strategy for Screening and Confirmation of 330 Pesticides in Ground- and Surface Water Using Liquid Chromatography Tandem Mass Spectrometry


 and
and
Abstract
:1. Introduction
2. Results
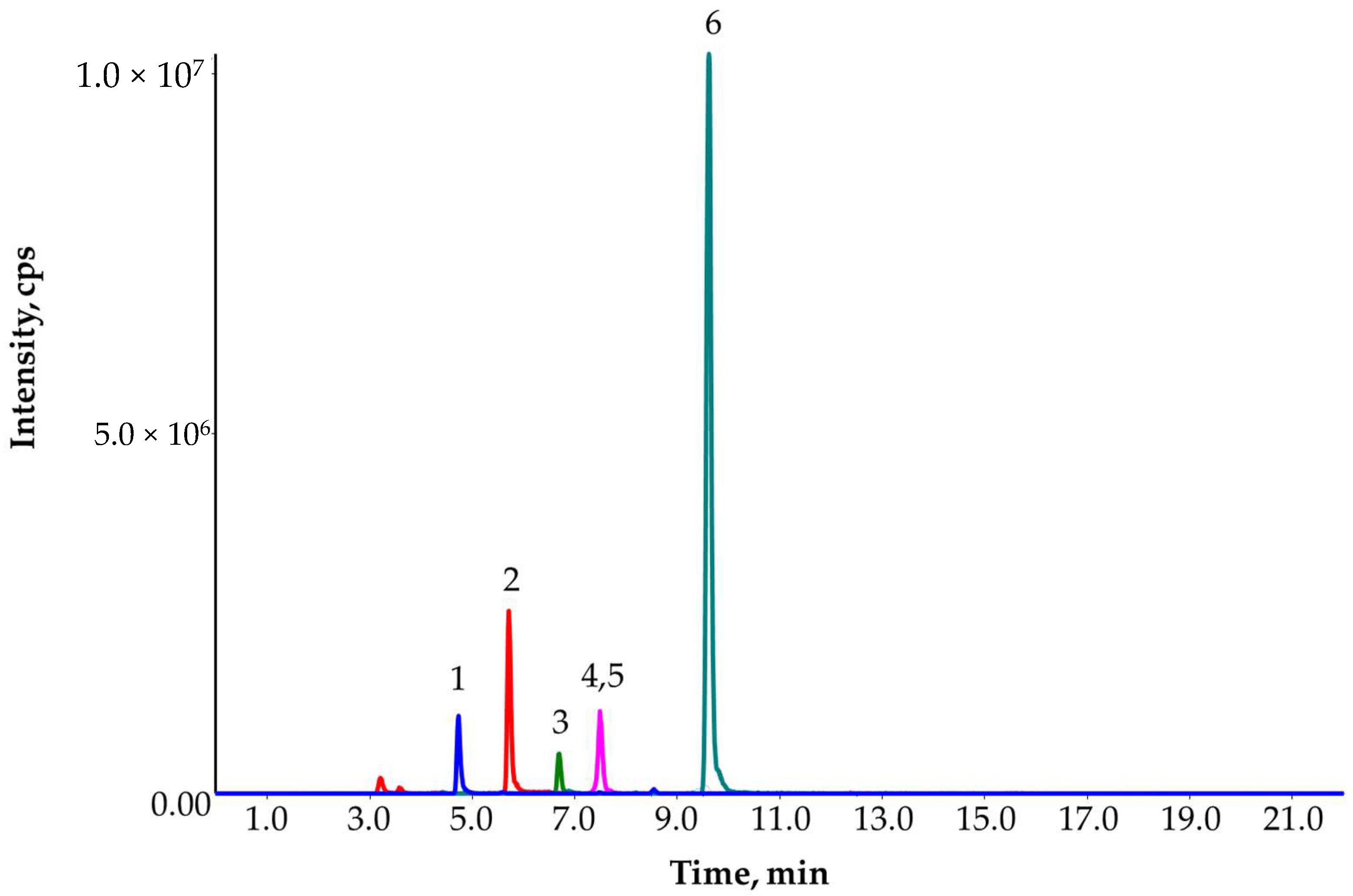
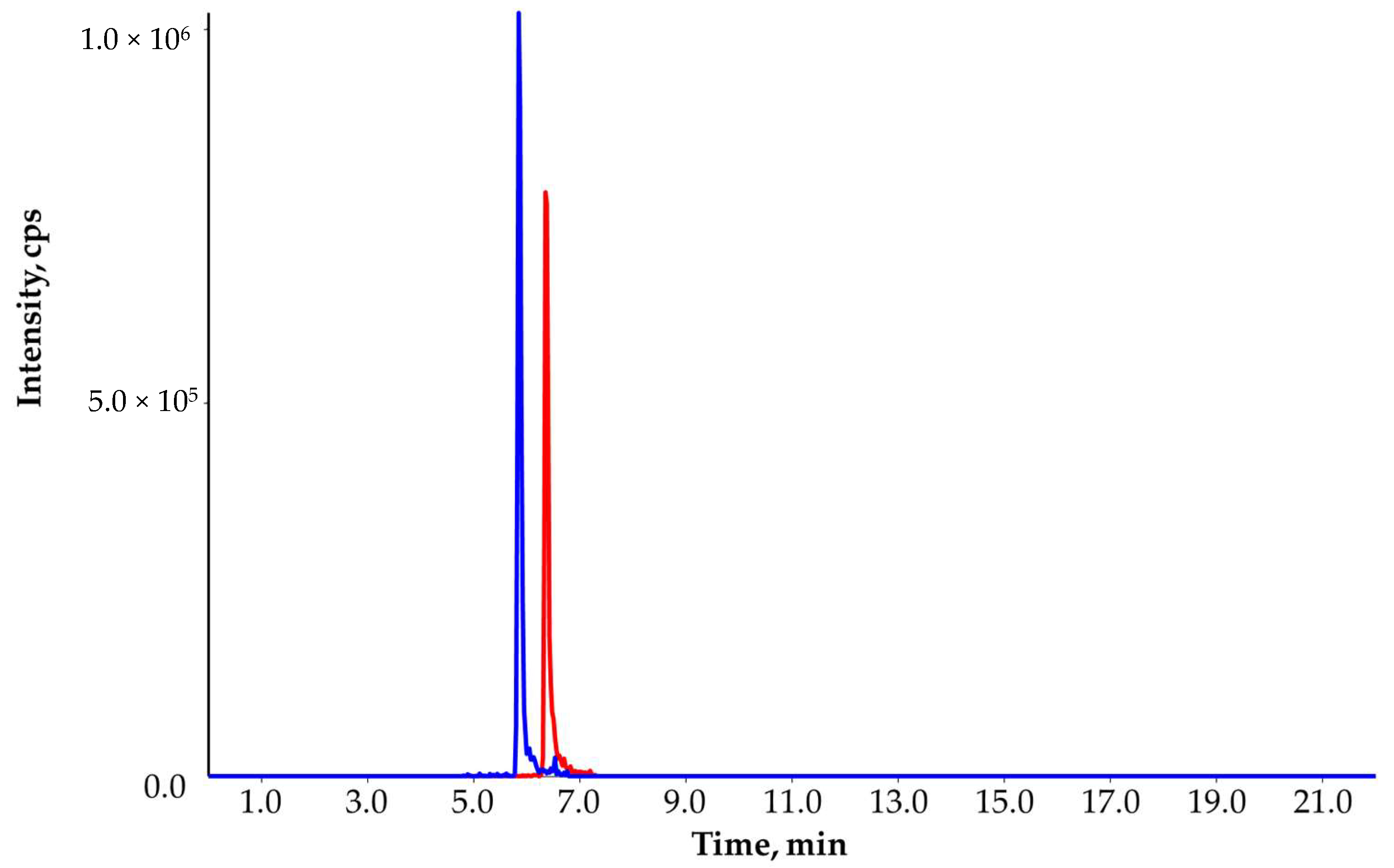
2.1. SPE-LC-MS/MS Method
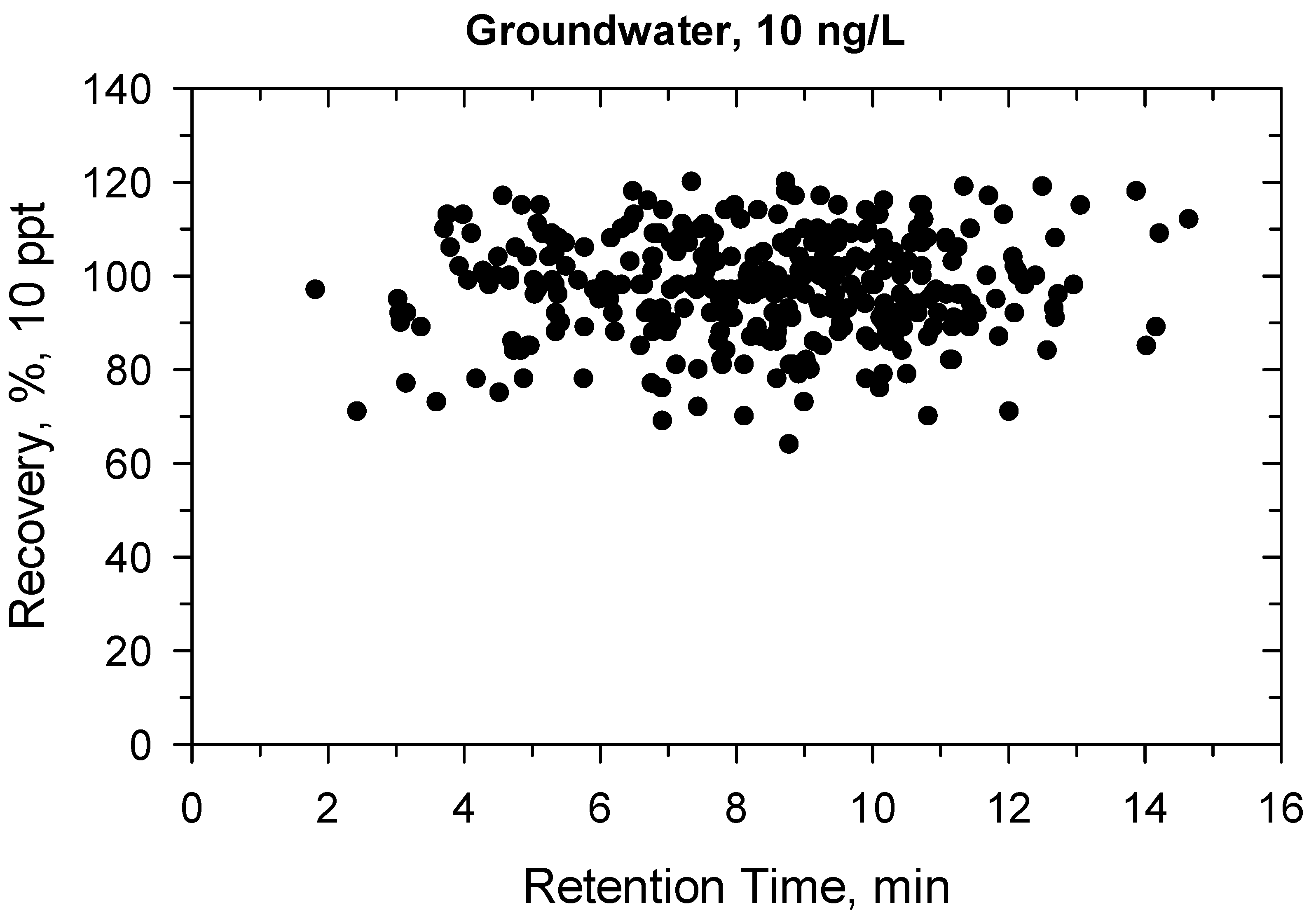
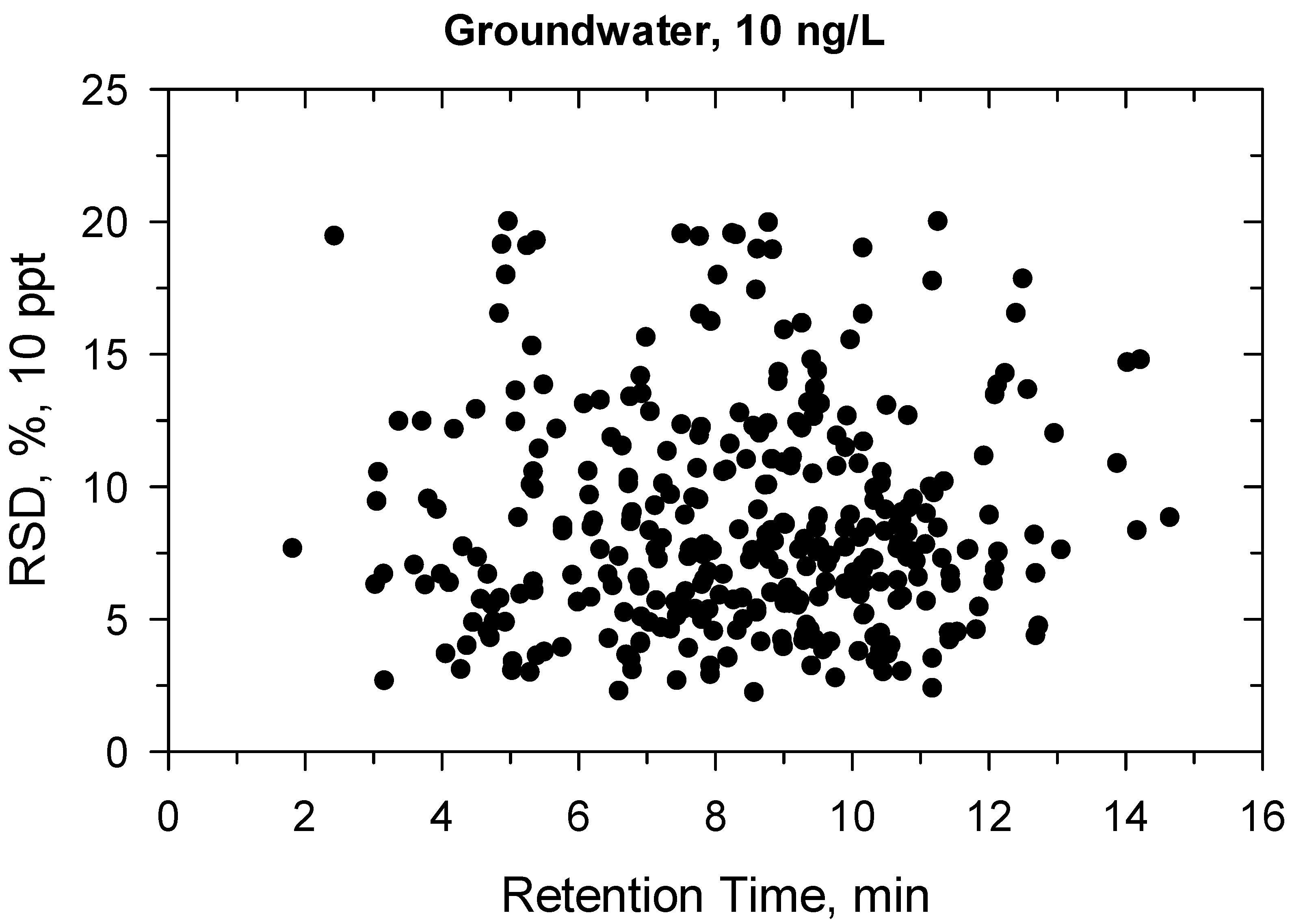
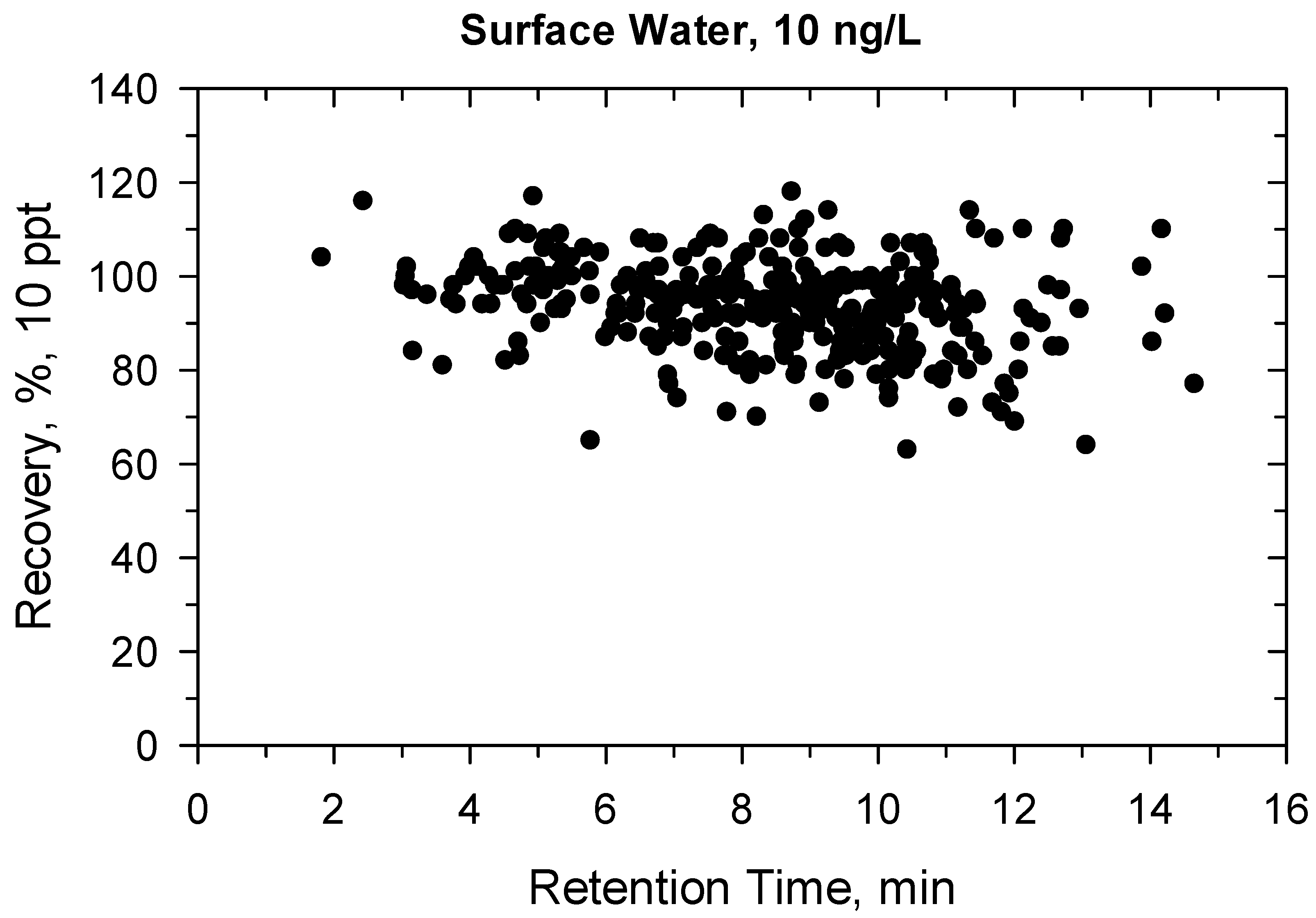
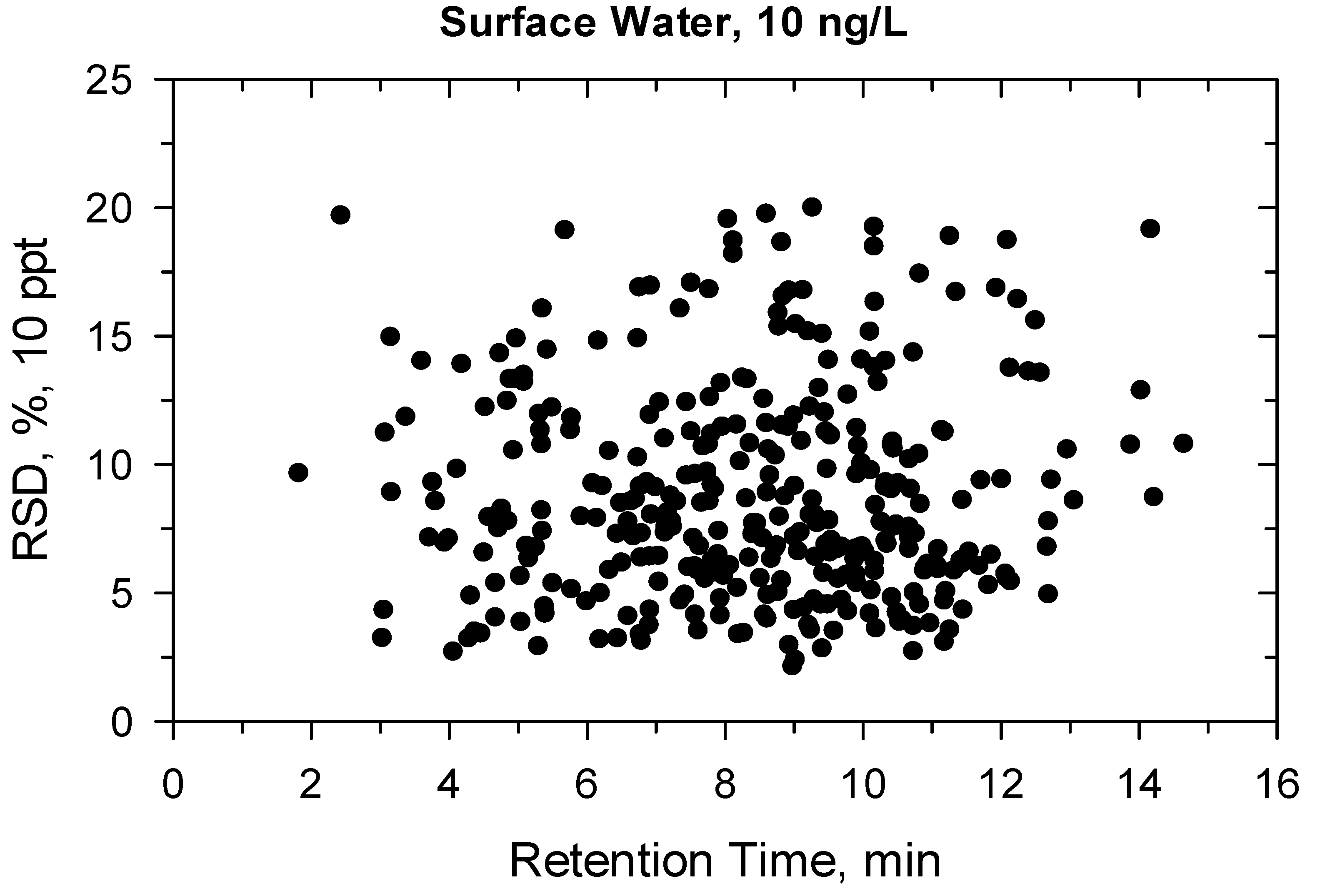
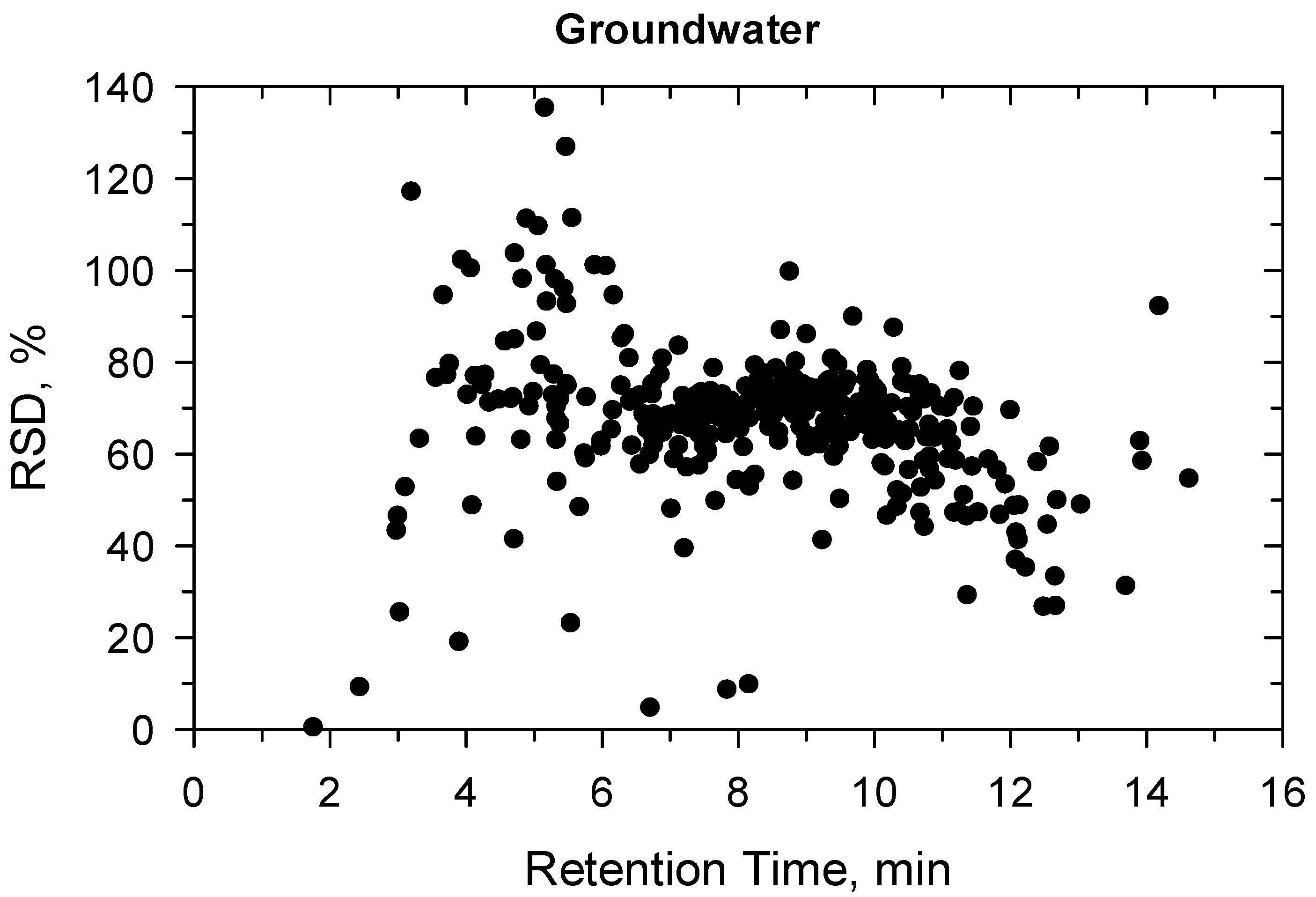
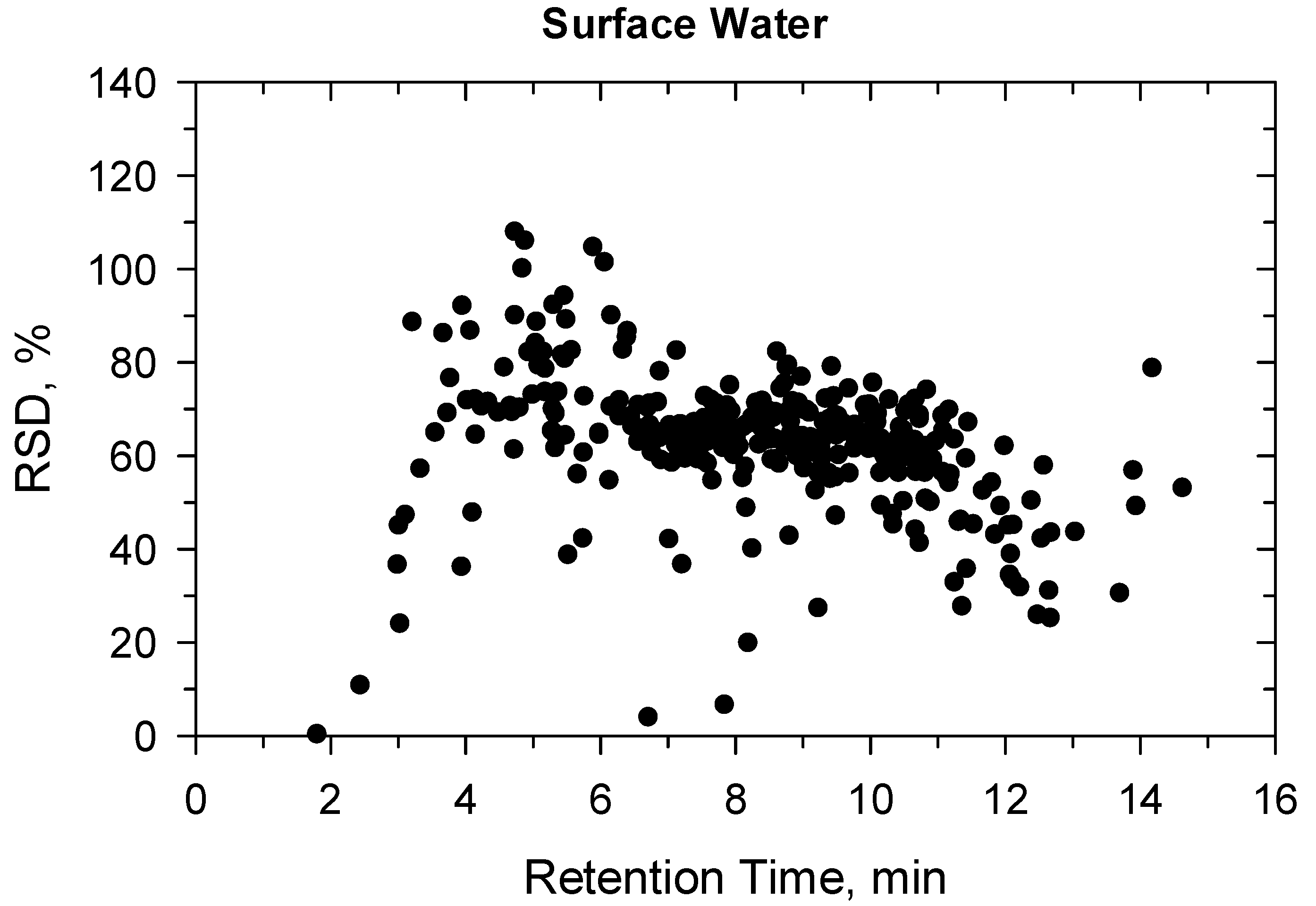
2.2. Method Validation
2.3. Proficiency Test
2.4. Automated SPE Pre-Concetration
2.5. Real Sample Analysis
3. Discussion
3.1. Method Development
3.2. Real Sample Analysis
4. Materials and Methods
4.1. Reagents and Samples
4.2. Instrumentation
4.3. Sample Preparation
4.4. LC-MS/MS Separation
4.5. Quantification
4.6. Validation
4.7. Automated SPE Enrichment
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Sample Availability
References
- De, A.; Bose, R.; Kumar, A.; Mozumdar, S. Worldwide Pesticide Use. In Targeted Delivery of Pesticides Using Biodegradable Polymeric Nanoparticles; Springer: New Delhi, India, 2014; pp. 5–6. [Google Scholar]
- Campanale, C.; Massarelli, C.; Losacco, D.; Bisaccia, D.; Triozzi, M.; Uricchio, V.F. The monitoring of pesticides in water matrices and the analytical criticalities: A review. Trends Anal. Chem. 2021, 144, 116423. [Google Scholar] [CrossRef]
- Khalid, S.; Shahid, M.; Murtaza, B.; Bibi, I.; Natasha; Naeem, M.A.; Niazi, N.K. A critical review of different factors governing the fate of pesticides in soil under biochar application. Sci. Total Environ. 2020, 711, 134645. [Google Scholar] [CrossRef] [PubMed]
- Veloo, K.V.; Ibrahim, N.A.S. Analytical Extraction Methods and Sorbents’ Development for Simultaneous Determination of Organophosphorus Pesticides’ Residues in Food and Water Samples: A Review. Molecules 2021, 26, 5495. [Google Scholar] [CrossRef]
- Syafrudin, M.; Kristanti, R.A.; Yuniarto, A.; Hadibarata, T.; Rhee, J.; Al-onazi, W.A.; Algarni, T.S.; Almarri, A.H.; Al-Mohaimeed, A.M. Pesticides in Drinking Water—A Review. Int. J. Environ. Res. Public Health 2021, 18, 468. [Google Scholar] [CrossRef] [PubMed]
- EU Pesticides Database. Available online: https://ec.europa.eu/food/plants/pesticides/eu-pesticides-database_hu (accessed on 8 February 2022).
- Sharma, A.; Kumar, V.; Shahzad, B.; Tanveer, M.; Sidhu, G.P.S.; Handa, N.; Kohli, S.K.; Yadav, P.; Bali, A.S.; Parihar, R.D. Worldwide pesticide usage and its impacts on ecosystem. SN Appl. Sci. 2019, 1, 1446. [Google Scholar] [CrossRef] [Green Version]
- El-Nahhal, I.; El-Nahhal, Y. Pesticide residues in drinking water, their potential risk to human health and removal options. J. Environ. Manag. 2021, 299, 113611. [Google Scholar] [CrossRef] [PubMed]
- Jayasiri, M.M.J.G.C.N.; Yadav, S.; Dayawansa, N.D.K.; Propper, C.R.; Kumar, V.; Singleton, G.R. Spatio-temporal analysis of water quality for pesticides and other agricultural pollutants in Deduru Oya river basin of Sri Lanka. J. Clean. Prod. 2022, 330, 129897. [Google Scholar] [CrossRef]
- Barbieri, M.V.; Postigo, C.; Monllor-Alcaraz, L.; Barceló, D.; de Alda, M.L. A reliable LC-MS/MS-based method for trace level determination of 50 medium to highly polar pesticide residues in sediments and ecological risk assessment. Anal. Bioanal. Chem. 2019, 411, 7981–7996. [Google Scholar] [CrossRef]
- Székács, A.; Mörtl, M.; Darvas, B. Monitoring pesticide residues in surface and ground water in Hungary: Surveys in 1990–2015. J. Chem. 2015, 2015, 717948. [Google Scholar] [CrossRef] [Green Version]
- European Parliament and Council. European Union Directive 2008/105/EC of the European Parliament and of the Council on environmental quality standards in the field of water policy. Off. J. Eur. Union 2008, L348, 84–97. Available online: https://eur-lex.europa.eu/eli/dir/2008/105/oj (accessed on 8 February 2022).
- European Parliament and Council. Directive 2013/39/EU of 12 August 2013 amending Directives 2000/60/EC and 2008/105/EC as regards priority substances in the field of water policy. Off. J. Eur. Union 2013, L226, 1–17. Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2013:226:0001:0017:EN:PDF (accessed on 8 February 2022).
- Greulich, K.; Alder, L. Fast multiresidue screening of 300 pesticides in water for human consumption by LC-MS/MS. Anal. Bioanal. Chem. 2008, 391, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Borrull, J.; Colom, A.; Fabregas, J.; Pocurull, E.; Borrull, F. A simple, fast method for the analysis of 20 contaminants of emerging concern in river water using large-volume direct injection liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2019, 411, 1601–1610. [Google Scholar] [CrossRef] [PubMed]
- Egli, M.; Hartmann, A.; Rapp Wright, H.; Ng, K.T.; Piel, F.B.; Barron, L.P. Quantitative Determination and Environmental Risk Assessment of 102 Chemicals of Emerging Concern in Wastewater-Impacted Rivers Using Rapid Direct-Injection Liquid Chromatography—Tandem Mass Spectrometry. Molecules 2021, 26, 5431. [Google Scholar] [CrossRef] [PubMed]
- Donato, F.F.; Martins, M.L.; Munaretto, J.S.; Prestes, O.D.; Adaime, M.B.; Zanella, R.J. Development of a Multiresidue Method for Pesticide Analysis in Drinking Water by Solid Phase Extraction and Determination by Gas and Liquid Chromatography with Triple Quadrupole Tandem Mass Spectrometry. J. Braz. Chem. Soc. 2015, 26, 2077–2087. [Google Scholar] [CrossRef]
- Liu, F.; Bischoff, G.; Pestemer, W.; Xu, W.; Kofoet, A. Multi-Residue Analysis of Some Polar Pesticides in Water Samples with SPE and LC–MS–MS. Chromatographia 2006, 63, 233–237. [Google Scholar] [CrossRef]
- Demoliner, A.; Caldas, S.S.; Costa, F.P.; Gonçalves, F.F.; Clementin, R.M.; Milani, M.R.; Primel, E.G. Development and validation of a method using SPE and LC-ESI-MS-MS for the determination of multiple classes of pesticides and metabolites in water samples. J. Braz. Chem. Soc. 2010, 21, 1424–1433. [Google Scholar] [CrossRef] [Green Version]
- Becker, R.W.; Araújo, D.S.; Sirtori, C.; Toyama, N.P.; Tavares, D.A.; Cordeiro, G.A.; Benassi, S.F.; Gossen, A.C.; do Amaral, B. Pesticides in surface water from Brazil and Paraguay cross-border region: Screening using LC-QTOF MS and correlation with land use and occupation through multivariate analysis. Microchem. J. 2021, 168, 106502. [Google Scholar] [CrossRef]
- Diaz, R.; Ibáñez, M.; Sancho, J.V.; Hernández, F. Qualitative validation of a liquid chromatography–quadrupole-time of flight mass spectrometry screening method for organic pollutants in waters. J. Chromatogr. A 2013, 1276, 47–57. [Google Scholar] [CrossRef]
- Mondal, R.; Kole, R.K. Monitoring and evaluation of pesticide residues in aquatic systems. In Sustainable Agriculture Reviews; Ahamed Inamuddin, M.I., Lichtfouse, E., Eds.; Springer: Cham, Switzerland, 2021; Volume 47, pp. 91–143. [Google Scholar] [CrossRef]
- Zheng, S.; Chen, B.; Qiu, X.; Chen, M.; Ma, Z.; Yu, X. Distribution and risk assessment of 82 pesticides in Jiulong River and estuary in South China. Chemosphere 2016, 144, 1177–1192. [Google Scholar] [CrossRef]
- Tölgyesi, Á.; Korozs, G.; Tóth, E.; Bálint, M.; Ma, X.; Sharma, V.K. Automation in quantifying phenoxy herbicides and bentazon in surface water and groundwater using novel solid phase extraction and liquid chromatography tandem mass spectrometry. Chemosphere 2022, 286, 131927. [Google Scholar] [CrossRef]
- Bexfield, L.M.; Belitz, K.; Lindsey, B.D.; Toccalino, P.L.; Nowell, L.H. Pesticides and Pesticide Degradates in Groundwater Used for Public Supply across the United States: Occurrence and Human-Health Context. Environ. Sci. Technol. 2021, 55, 362–372. [Google Scholar] [CrossRef]
- EN 15662:2018; Foods of Plant Origin—Multimethod for the Determination of Pesticide Residues Using GC- and LC-Based Analysis Following Acetonitrile Extraction/Partitioning and Clean-Up by Dispersive SPE—Modular QuEChERS-Method. Available online: https://standards.iteh.ai/catalog/standards/cen/167a30bc-edf9-4cf8-b96b-cabd932f2f02/en-15662-2018 (accessed on 8 February 2022).
- Sereshti, H.; Seraj, M.; Soltani, S.; Nodeh, H.R.; AliAbadi, M.H.S.; Taghizadeh, M. Development of a sustainable dispersive liquid–liquid microextraction based on novel hydrophobic and hydrophilic natural deep eutectic solvents for the analysis of multiclass pesticides in water. Microchem. J. 2022, 175, 107226. [Google Scholar] [CrossRef]
- Dąbrowski, L. Review of use of keepers in solvent evaporation procedure during the environmental sample analysis of some organic pollutants. TrAC Trends Anal. Chem. 2016, 80, 507–516. [Google Scholar] [CrossRef]
- Kapsi, M.; Tsoutsi, C.; Albanis, T. Simple analytical methodology based on solid phase extraction for monitoring pesticide residues in natural waters. MethodsX 2020, 7, 101011. [Google Scholar] [CrossRef] [PubMed]
- Nakhjavan, B.; Bland, J.; Khosravifard, M. Optimization of a Multiresidue Analysis of 65 Pesticides in Surface Water Using Solid-Phase Extraction by LC-MS/MS. Molecules 2021, 26, 6627. [Google Scholar] [CrossRef] [PubMed]
- Blanchoud, H.; Alliot, F.; Chen, N.; Valdes, D. Rapid SPE–LC MS/MS analysis for atrazine, its by-products, simazine and S metolachlor in groundwater samples. MethodsX 2020, 7, 100824. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-Y.; Fodjo, E.K.; Kong, C.; Yu, H.-J. Multi-residue screening of pesticides in aquaculture waters through ultra-high-performance liquid chromatography-Q/orbitrap mass spectrometry. Water 2020, 12, 1238. [Google Scholar] [CrossRef]
- Shamsipur, M.; Yazdanfar, N.; Ghambarian, M. Combination of solid-phase extraction with dispersive liquid-liquid microextraction followed by GC-MS for determination of pesticide residues from water, milk, honey and fruit juice. Food Chem. 2016, 204, 289–297. [Google Scholar] [CrossRef]
- EPA. Method 1658: The Determination of Phenoxy-Acid Herbicides in Municipal and Industrial Wastewater. 1993. Available online: https://www.epa.gov/sites/production/files/2015-10/documents/method_1658_1993.pdf (accessed on 8 February 2022).
- Pacheco-Fernández, I.; Pino, V. Green solvents in analytical chemistry. Curr. Opin. Green Sustain. Chem. 2019, 18, 42–50. [Google Scholar] [CrossRef]
- EPA Method 3535A (SW-846). Solid-Phase Extraction (SPE). Available online: https://www.epa.gov/esam/epa-method-3535a-sw-846-solid-phase-extraction-spe (accessed on 8 February 2022).
- EPA. Method 8321B: Solvent-Extractable Nonvolatile Compounds by High-Performance Liquid Chromatography/Thermospray/Mass Spectrometry (HPLC/TS/MS) or Ultraviolet (UV) Detection. 2007. Available online: https://www.epa.gov/sites/production/files/2015-12/documents/8321b.pdf (accessed on 8 February 2022).
- Tölgyesi, Á.; Tóth Kovács, B.; Tóth, E.; Simon, A.; Bálint, M.; Sharma, V.K. Unexpected sensitivity enhancement in analyzing aflatoxin M1 in food using LC-IDMS separation. Microchem. J. 2022. submitted. [Google Scholar]
- Kaur, S.; Kumar, V.; Chawla, M.; Cavallo, L.; Poater, A.; Upadhyay, N. Pesticides Curbing Soil Fertility: Effect of Complexation of Free Metal Ions. Front. Chem. 2017, 5, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CEN/TS 17061:2017; Foodstuffs-Guidelines for the Calibration and Quantitative Determination of Pesticide Residues and Organic Contaminants Using Chromatographic Methods. Available online: https://standards.iteh.ai/catalog/standards/cen/d4e843c5-a110-4e6b-a7c6-652b5a169241/cen-ts-17061-2017 (accessed on 8 February 2022).
- SANTE/12682/2019; Guidance Document on Analytical Quality Control and Method Validation Procedures for Pesticides Residues Analysis in Food and Feed, Supersedes Document No.SANTE/2017/11813. Implemented by 1 January 2020. Available online: https://ec.europa.eu/food/sites/food/files/plant/docs/pesticides_mrl_guidelines_wrkdoc_2019-12682.pdf (accessed on 8 February 2022).
- Lepom, P.; Brown, B.; Hanke, G.; Loos, R.; Quevauviller, P.; Wollgast, J. Needs for reliable analytical methods for monitoring chemical pollutants in surface water under the European Water Framework Directive. J. Chromatogr. A 2009, 1216, 302–315. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Sample | Result (ng/L) | Deviation from the Assigned Value% | Permitted Deviation% | Evaluation |
|---|---|---|---|---|---|
| Atrazine | 1 | 51.2 | −5 | ±35 | Satisfactory |
| Atrazine | 2 | 99.6 | +3 | ±35 | Satisfactory |
| Desethyl atrazine | 1 | 51.7 | +5 | ±35 | Satisfactory |
| Desethyl atrazine | 2 | 63.0 | −1 | ±35 | Satisfactory |
| Desizopropyl atrazine | 1 | 54.6 | +20 | ±35 | Satisfactory |
| Metazachlor | 1 | 27.0 | −30 | ±35 | Satisfactory |
| Metazachlor | 2 | 74.0 | −21 | ±35 | Satisfactory |
| Metolachlor | 1 | 55.0 | +1 | ±35 | Satisfactory |
| Metolachlor | 2 | 183 | +8 | ±35 | Satisfactory |
| Simazine | 1 | 50.0 | −8 | ±35 | Satisfactory |
| Simazine | 2 | 53.0 | −5 | ±35 | Satisfactory |
| Sample Type | Number of Compounds Identified under 10 ng/L (Screening) | Confirmed Concentration of Compounds under 10 ng/L | Identified Compound above 10 ng/L (Screening) | Confirmed Concentration Range and Sum of Compounds above 10 ng/L | Total Number of Confirmed Compounds | Sum Concentration of All Compounds (ng/L) |
|---|---|---|---|---|---|---|
| Groundwater #1 | 3 | 1.50–7.55 | - | - | 3 | 11.8 |
| Surface water #1 | 2 | 2.25–4.33 | Carbendazim, Thiabendazole | 12.62–15.35 (∑28.0 ng/L) | 4 | 34.5 |
| Surface water #2 | 4 | 1.79–8.46 | - | - | 4 | 19.5 |
| Surface water #3 | 11 | 1.69–9.52 | Acetamiprid, Boscalid, Carbendazim, Fenhexamid, Imidacloprid, Penconazole, Terbuthylazine-desethyl | 12.2–36.3 (∑163 ng/L) | 18 | 213 |
| Surface water #4 | 14 | 1.93–5.61 | Azoxystrobin, Imidacloprid, Metolachlor, Thiabendazole | 11.2–42.4 (∑92.9 ng/L) | 18 | 159 |
| Surface water #5 | 4 | 1.91–7.87 | - | - | 4 | 15.7 |
| Surface water #6 | 9 | 1.07–9.28 | - | - | 9 | 30.0 |
| Surface water #7 | 6 | 1.42–4.63 | Carbendazim, Thiabendazole | 14.1–16.4 (∑30.5 ng/L) | 8 | 45.8 |
| Surface water #8 | 5 | 1.30–3.75 | Carbendazim, Thiabendazole | 10.3–10.4 (∑20.7 ng/L) | 7 | 31.3 |
| Surface water #9 | 11 | 1.95–8.27 | Atrazine-desethyl, Imidacloprid, Thiabendazole | 10.9–43.9 (∑85.8 ng/L) | 14 | 131 |
| Surface water #10 | 6 | 1.00–4.29 | Carbendazim, Thiabendazole | 12.0–16.8 (∑28.8 ng/L) | 8 | 45.2 |
| Surface water #11 | 14 | 1.31–9.39 | Azoxystrobin, Boscalid, Carbendazim, Imidacloprid, Isoproturon, Metolachlor, Tebuconazole, Thiabendazole | 10.5–37.1 (∑146 ng/L) | 8 | 219 |
| Surface water #12 | 6 | 1.21–3.58 | Carbendazim, Thiabendazole | 10.1–10.1 (∑20.2 ng/L) | 8 | 34.7 |
| Surface water #13 | 6 | 1.00–4.37 | Carbendazim, Thiabendazole | 15.0–16.3 (∑31.3 ng/L) | 8 | 46.1 |
| Surface water #14 | 8 | 1.24–7.45 | Atrazine-desethyl | 31.9 (∑31.3 ng/L) | 9 | 58.4 |
| Surface water #15 | 5 | 1.07–9.23 | Carbendazim | 10.3 (∑10.3 ng/L) | 6 | 26.2 |
| Surface water #16 | 7 | 1.10–4.58 | Carbendazim, Thiabendazole | 10.6–12.0 (∑22.6 ng/L) | 9 | 36.5 |
| Surface water #17 | 4 | 1.16–3.45 | Carbendazim, Thiabendazole | 10.2–12.3 (∑22.5 ng/L) | 6 | 30.8 |
| Surface water #18 | 129 | 1.04–4.94 | Carbendazim, Thiabendazole | 18.8–14.4 (∑33.2 ng/L) | 131 | 245 |
| Surface water #19 | 8 | 1.07–4.18 | Carbendazim, Thiabendazole | 11.3–13.8 (∑25.0 ng/L) | 10 | 41.5 |
| Number of Compounds Analyzed | Sample Preparation | LOQ (ng/L) | Ref. |
|---|---|---|---|
| 300 | direct injection | 100–1000 | [14] |
| 20 | direct injection | 0.5–2.0 | [15] |
| 102 | direct injection | 10–700 | [16] |
| 7 | SPE | 10 | [18] |
| 18 | SPE | 4–100 | [19] |
| 43 | SPE | 100–1000 | [20] |
| 150 | SPE | 100–1000 | [21] |
| 12 | SPE | 5–99 | [29] |
| 65 | SPE | 1.67–5.37 | [30] |
| 6 | on-line SPE | 1–15 | [31] |
| 22 | SPE | 2–1000 | [32] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tóth, E.; Tölgyesi, Á.; Simon, A.; Bálint, M.; Ma, X.; Sharma, V.K. An Alternative Strategy for Screening and Confirmation of 330 Pesticides in Ground- and Surface Water Using Liquid Chromatography Tandem Mass Spectrometry. Molecules 2022, 27, 1872. https://doi.org/10.3390/molecules27061872
Tóth E, Tölgyesi Á, Simon A, Bálint M, Ma X, Sharma VK. An Alternative Strategy for Screening and Confirmation of 330 Pesticides in Ground- and Surface Water Using Liquid Chromatography Tandem Mass Spectrometry. Molecules. 2022; 27(6):1872. https://doi.org/10.3390/molecules27061872
Chicago/Turabian StyleTóth, Edgár, Ádám Tölgyesi, Andrea Simon, Mária Bálint, Xingmao Ma, and Virender K. Sharma. 2022. "An Alternative Strategy for Screening and Confirmation of 330 Pesticides in Ground- and Surface Water Using Liquid Chromatography Tandem Mass Spectrometry" Molecules 27, no. 6: 1872. https://doi.org/10.3390/molecules27061872
APA StyleTóth, E., Tölgyesi, Á., Simon, A., Bálint, M., Ma, X., & Sharma, V. K. (2022). An Alternative Strategy for Screening and Confirmation of 330 Pesticides in Ground- and Surface Water Using Liquid Chromatography Tandem Mass Spectrometry. Molecules, 27(6), 1872. https://doi.org/10.3390/molecules27061872