
Analytical Methods for the Determination of 90Sr and 239,240Pu in Environmental Samples


Abstract
:1. Introduction
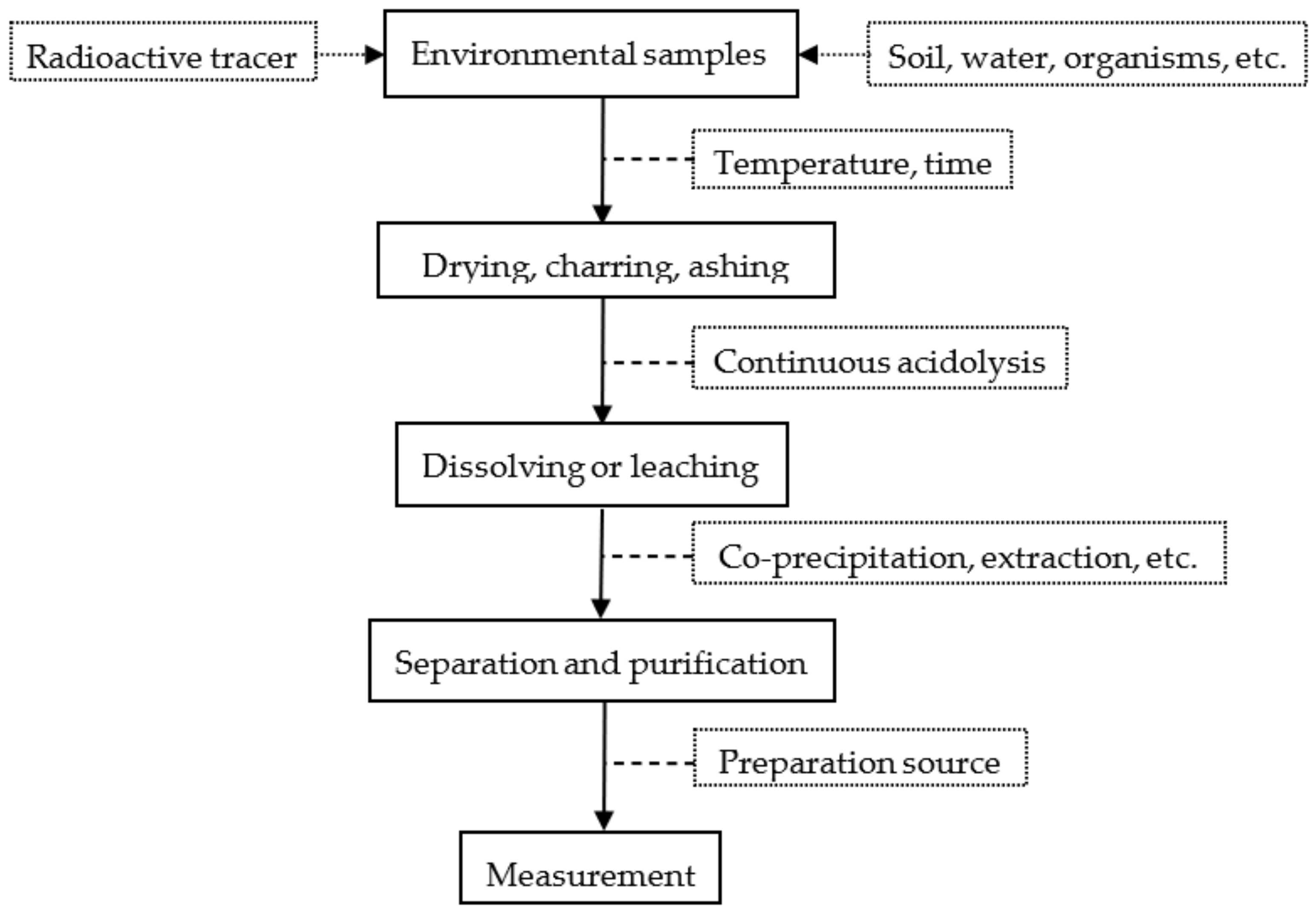
2. Sample Preparation
2.1. Sample Pretreatment
2.2. Chemical Separation
2.2.1. Separation Methods of 90Sr in Environmental Samples
2.2.2. Separation Methods of Pu in Environmental Samples
3. Measurement Methods of 239Pu, 240Pu, and 90Sr
3.1. Radioactivity Measurement Methods
3.2. Mass Spectrometry Methods
3.2.1. Measurement Method of 90Sr
3.2.2. Mass Spectrometry Measurement Methods of 239Pu and 240Pu
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Feuerstein, J.; Boulyga, S.F.; Galler, P.; Stingeder, G.; Prohaska, T. Determination of 90Sr in soil samples using inductively coupled plasma mass spectrometry equipped with dynamic reaction cell (ICP-DRC-MS). J. Environ. Radioact. 2008, 99, 1764–1769. [Google Scholar] [CrossRef]
- Mole, R.H. Working with plutonium. Lancet 1975, 305, 701–702. [Google Scholar] [CrossRef]
- Langham, W.H.; Healy, J. Maximum permissible body burdens and concentrations of plutonium: Biological basis and history of development. In Uranium-Plutonium Transplutonic Elements; Springer: Berlin/Heidelberg, Germany, 1973; Volume 36, pp. 569–592. [Google Scholar]
- Aliyu, A.S.; Evangeliou, N.; Mousseau, T.A.; Wu, J.; Ramli, A.T. An overview of current knowledge concerning the health and environmental consequences of the Fukushima Daiichi Nuclear Power Plant (FDNPP) accident. Environ. Int. 2015, 85, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Runde, W. The chemical interactions of actinides in the environment. Alamos Sci. 2000, 26, 392–411. [Google Scholar]
- Li, M.F.; Luo, D.L.; Weng, H.S.; Zhang, Z.; Yu, J.Y. The transfer coefficients of Ba, Sr, and Zr in Guangdong cabbage. J. Sun Yat-sen Univ. Nat. Sci. 1997, 36, 109–112. [Google Scholar]
- Weng, C.W.; Huang, H.S.; Xu, C.X. Study on the accumulation effect of 11 heavy metals in vegetables. Food Res. Dev. 2018, 39, 194–200. [Google Scholar]
- Cai, F.L.; Chen, Y.; Huang, L.Y. Research on Mytilus viridis linnaeus as indicator organisms for 90Sr pollution in sea area. J. Oceanogr. 1990, 12, 261–264. [Google Scholar]
- Neck, V.; Altmaier, M.; Seibert, A.; Yun, J.I.; Marquardt, C.M.; Fanghanel, T. Solubility and redox reactions of Pu(IV) hydrous oxide: Evidence for the formation of PuO2+x(s, hyd). Radiochim. Acta 2007, 95, 137d9–207. [Google Scholar] [CrossRef]
- Lee, J.H.; Hossnera, L.R.; Attrep, M.; Kung, K.S. Uptake and translocation of plutonium in two plant species using hydroponics. Environ. Pollut. 2002, 117, 61–68. [Google Scholar] [CrossRef]
- Tawussi, F.; Gupta, D.; Muhr-Ebert, E.; Schneider, S.; Bister, S.; Walther, C. Uptake of Plutonium-238 into Solanum tuberosum L. (potato plants) in presence of complexing agent EDTA. J. Environ. Radioact. 2017, 178-179, 186–192. [Google Scholar] [CrossRef]
- Kenna, T.C. Determination of plutonium isotopes and neptunium-237 in environmental samples by inductively coupled plasma mass spectrometry with total sample dissolution. J. Anal. At. Spectrom. 2002, 17, 1471–1479. [Google Scholar] [CrossRef]
- Mitchell, P.I.; Vintro, L.L.; Dahlgard, H.; Gasco, C.; Sanchez-Cabeza, J.A. Perturbation in the 240Pu239Pu global fallout ratio in local sediments following the nuclear accidents at Thule (Greenland) and Palomares (Spain). Sci. Total Environ. 1997, 202, 147–153. [Google Scholar] [CrossRef]
- Erdmann, N.; Herrmann, G.; Huber, G.; Kohler, S.; Kratz, J.V.; Mansel, A.; Nunnemann, M.; Passler, G.; Trautmann, N.; Turchin, A.; et al. Resonance Ionization Mass Spectroscopy for Trace Determination of Plutonium in Environmental Samples. Fresenius’ J. Anal. Chem. 1997, 359, 378–381. [Google Scholar] [CrossRef]
- Eriksson, M.; Lindahl, P.; Roos, P.; Dahlgaard, H.; Holm, E. U, Pu, and Am nuclear signatures of the thule hydrogen bomb debris. Environ. Sci. Technol. 2008, 42, 4717–4722. [Google Scholar] [CrossRef]
- Krey, P.W.; Hardy, E.P.; Pachucki, C.; Rourke, F.; Coluzza, J.; Benson, W.K. Mass Isotopic Composition of Global Fall-Out Plutonium in Soil. In Transuranium Nuclides in the Environment, Proceedings of the Symposium on Transuranium Nuclides in the Environment, San Francisco, CA, USA, 17–21 November 1975; International Atomic Energy Agency: Vienna, Austria, 1976; pp. 671–678. [Google Scholar]
- Igarashi, J.; Zheng, J.; Zhang, Z.; Ninomiya, K.; Satou, Y.; Fukuda, M.; Ni, Y.; Aono, T.; Shinohara, A. First determination of Pu isotopes (239Pu, 240Pu and 241Pu) in radioactive particles derived from Fukushima Daiichi Nuclear Power Plant accident. Sci. Rep. 2019, 9, 11807. [Google Scholar] [CrossRef] [Green Version]
- Muramatsu, Y.; Rühm, W.; Yoshida, S.; Tagami, K.; Uchida, S.; Wirth, E. Concentrations of 239Pu and 240Pu and their isotopic ratios determined by ICP-MS in soils collected from the Chernobyl 30-km zone. Environ. Sci. Technol. 2000, 34, 2913–2917. [Google Scholar] [CrossRef]
- Vonderheide, A.P.; Zoriy, M.V.; Izmer, A.V.; Pickhardt, C.; Caruso, J.A.; Ostapczuk, P.; Hille, R.; Becker, J.S. Determination of 90Sr at ultratrace levels in urine by ICP-MS. J. Anal. At. Spectrom. 2004, 19, 675–680. [Google Scholar] [CrossRef] [Green Version]
- Helal, A.I.; Zahran, N.F.; Amr, M.A.; El-Lateef, A.M.A.; Bashter, I.I.; Mohsen, H.T.; Abbas, Y. Ultratrace and isotope ratios analyses of some radionuclides by ICP-MS. Radiochim. Acta 2004, 92, 369–374. [Google Scholar] [CrossRef]
- Amr, M.A.; Abdel-Lateef, A.M. Comparing the capability of collision/reaction cell quadrupole and sector field inductively coupled plasma mass spectrometers for interference removal from 90Sr, 137Cs, and 226Ra. Int. J. Mass Spectrom. 2011, 299, 184–190. [Google Scholar] [CrossRef]
- Goldstein, S.; Price, A.; Hinrichs, K.; Lamont, S.; Nunn, A.; Amato, R.; Cardon, A.; Gurganus, D. High-precision measurement of U-Pu-Np-Am concentrations and isotope ratios in environmental reference materials by mass spectrometry. J. Environ. Radioact. 2021, 237, 106689. [Google Scholar] [CrossRef]
- Takagai, Y.; Furukawa, M.; Kameo, Y.; Suzuki, K. Sequential inductively coupled plasma quadrupole mass-spectrometric quantification of radioactive strontium-90 incorporating cascade separation steps for radioactive contamination rapid survey. Anal. Methods 2014, 6, 355–362. [Google Scholar] [CrossRef]
- Florou, H.; Kehagia, K.; Chaloulou, C.; Koukouliou, V.; Lykomitrou, C. Determination of radionuclides in Mytilus galloprovicialis by Alpha and Gamma-Spectroscopy. Mediterr. Mar. Sci. 2012, 5, 117–123. [Google Scholar] [CrossRef] [Green Version]
- Li, K.X.; Qin, S.C. Pretreatment of biological samples in the background investigation of radiation environment. J. Nav. Med. 2005, 26, 201–203. [Google Scholar]
- GB/T 16145—2020; Gamma Spectrometry Method of Analysing Radionuclides in Biological Samples. National Standardization Administration: Beijing, China, 2020.
- Ma, Y.; Tang, X.Z.; Ding, T.T. The application of microwave ashing analysis in the pretreatment of oil metal content. Anal. Test. Technol. Instrum. 2010, 16, 206–208. [Google Scholar]
- Bao, M.M.; Kang, J.W.; Jiang, D.C.; Li, P.; Wang, L.Y.; Zhu, S.W. Improvement of powdery sample detection dry ashing pretreatment. Light Ind. Technol. Food Biol. 2013, 12, 4–5. [Google Scholar]
- GB 14883.1—2016; General Rules for the Inspection of Radioactive Substances in Food. National Health and Family Planning: Beijing, China, 2017.
- Maxwell, S.L.; Culligan, B.K.; Hutchison, J.B. Rapid fusion method for determination of plutonium isotopes in large rice samples. J. Radioanal. Nucl. Chem. 2013, 298, 1367–1374. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Comparison of different digestion methods-inductively coupled plasma mass spectrometry for the determination of 15 metal elements in black tea. J. Tea 2019, 60, 151–155. [Google Scholar]
- Grahek, Z.; Dulanska, S.; Karanovic, G.; Coha, I.; Tucakovic, I.; Nodilo, M.; Matel, L. Comparison of different methodologies for the 90Sr determination in environmental samples. J. Environ. Radioact. 2018, 181, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Bu, W.; Zheng, J.; Liu, X.; Long, K.; Hu, S.; Uchida, S. Mass spectrometry for the determination of fission products 135Cs, 137Cs and 90Sr: A review of methodology and applications. Spectrochim. Acta B At. Spectrosc. 2016, 119, 65–75. [Google Scholar] [CrossRef]
- Amr, M.; Helal, A.-F.; Al-Kinani, A.T.; Balakrishnan, P. Ultra-trace determination of 90Sr, 137Cs, 238Pu, 239Pu, and 240Pu by triple quadruple collision/reaction cell-ICP-MS/MS: Establishing a baseline for global fallout in Qatar soil and sediments. J. Environ. Radioact. 2016, 153, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Mola, M.; Avivar, J.; Nieto, A.; Peñalver, A.; Aguilar, C.; Ferrer, L.; Cerdà, V.; Borrull, F. Determination of 90Sr and 210Pb in sludge samples using a LOV-MSFIA system and liquid scintillation counting. Appl. Radiat. Isot. 2014, 86, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Hurtado-Bermudez, S.; Mas, J.L.; Villa-Alfageme, M. A sequential determination of 90Sr and 210Po in food samples. Food Chem. 2017, 229, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.; Elahi, S.; Lee, K.; Fairman, B. A rapid and accurate method for the determination of plutonium in food using magnetic sector ICP-MS with an ultra-sonic nebuliser and ion chromatography. J. Environ. Monit. 2003, 5, 175–179. [Google Scholar] [CrossRef]
- Holm, E.; Ballestra, S.; Fukai, R. A method for ion-exchange separation of low levels of americium in environmental materials. Talanta 1979, 26, 791–794. [Google Scholar] [CrossRef]
- Hamato, A. An anion exchange method for the determination of 241Am and plutonium in environmental and biological samples. J. Radioanal. Nucl. Chem. 1982, 75, 265. [Google Scholar] [CrossRef]
- Matishov, G.G.; Matishov, D.G.; Namjatov, A.A.; Carroll, J.; Dahle, S. Anthropogenic radionuclides in Kola and Motovsky bays of the barents Sea, Russia. J. Environ. Radioact. 1999, 43, 77–88. [Google Scholar] [CrossRef]
- Lee, Y.K.; Bakhtiar, S.N.; Akbarzadeh, M.; Lee, J.S. Sequential isotopic determination of strontium, thorium, plutonium, uranium, and americium in bioassay samples. J. Radioanal. Nucl. Chem. 2000, 243, 525. [Google Scholar] [CrossRef]
- Heldal, H.E.; Varskog, P.; Føyn, L. Distribution of selected anthropogenic radionuclides (137Cs, 238Pu, 239,240Pu and 241Am) in marine sediments with emphasis on the Spitsbergene-Bear Island Area. Sci. Total Environ. 2002, 293, 233–245. [Google Scholar] [CrossRef]
- Qiao, J.X.; Hou, X.L.; Miro, M.; Roos, P. Determination of plutonium isotopes in waters and environmental solids: A Review. Anal. Chim. Acta 2009, 652, 66–84. [Google Scholar] [CrossRef] [Green Version]
- Vajda, N.; Kim, C.K. Determination of radiostrontium isotopes: A review of analytical methodology. Appl. Radiat. Isot. 2010, 68, 2306–2326. [Google Scholar] [CrossRef]
- Marzo, G.A. Atmospheric transport and deposition of radionuclides released after the Fukushima Daichi accident and resulting effective dose. Atmos. Environ. 2014, 94, 709–722. [Google Scholar] [CrossRef]
- Tayeb, M.; Dai, X.; Sdraulig, S. Rapid and simultaneous determination of Strontium-89 and Strontium-90 in seawater. J. Environ. Radioact. 2016, 153, 214–221. [Google Scholar] [CrossRef]
- Bojanowski, R.; Knapinska-Skiba, D. Determination of low level 90Sr in environmental samples: A novel approach to the classical method. J. Radioanal. Nucl. Chem. 1990, 138, 207–218. [Google Scholar] [CrossRef]
- Chobola, R.; Mell, P.; Daroczi, L.; Vincze, A. Rapid determination of radiostrontium isotopes in samples of NPP origin. J. Radioanal. Nucl. Chem. 2006, 267, 297–304. [Google Scholar] [CrossRef]
- Rao, D.D.; Mehendarge, S.T.; Chandramouli, S.; Hegde, A.G.; Mishra, U.C. Application of Cherenkov radiation counting for determination of 90Sr in environmental samples. J. Environ. Radio. 2000, 48, 49–57. [Google Scholar] [CrossRef]
- Tera, F.; Morrison, G.H. Radiochemical separations by isotopic ion exchange. Anal. Chem. 1966, 38, 959–964. [Google Scholar] [CrossRef]
- Kimura, T.; Iwashima, K.; Ishimori, T.; Hamada, T. Separation of strontium-89 and -90 from calcium in milk with a macrocyclic ether. Anal. Chem. 1979, 51, 1113–1116. [Google Scholar] [CrossRef]
- Pawlak, D.; Parus, J.; Dziel, T.; Muklaniwicz, A.; Mikolajczak, R. Determination of 90Sr traces in medical 90Y after separation on DGA column. Talanta 2013, 114, 1–4. [Google Scholar] [CrossRef]
- Horwitz, E.P.; McAlister, D.R.; Bond, A.H.; Barrans, R.E. Novel Extraction of Chromatographic Resins Based on Tetraalkyldiglycolamides: Characterization and Potential Applications. Solvent Extr. Ion Exch. 2005, 23, 319–344. [Google Scholar] [CrossRef]
- Horwitz, E.P.; Chiarizia, R.; Dietz, M.L. A novel strontium-selective extraction chromatographic resin. Solvent Extr. Ion Exch. 1992, 10, 313–336. [Google Scholar] [CrossRef]
- Taylor, V.F.; Evans, R.D.; Cornett, R.J. Determination of 90Sr in contaminated environmental samples by tuneable bandpass dynamic reaction cell ICP–MS. Anal. Bioanal. Chem. 2007, 387, 343–350. [Google Scholar] [CrossRef]
- Odoh, S.O.; Bylaska, E.J.; de Jong, W.A. Coordination and hydrolysis of plutonium ions in aqueous solution using Car–Parrinello molecular dynamics free energy simulations. J. Phys. Chem. A 2013, 117, 12256–12267. [Google Scholar] [CrossRef]
- Horwitz, E.P.; Dietz, M.L.; Chiarizia, R.; Diamond, H.; Maxwell, S.L.; Nelson, M.R. Separation and preconcentration of actinides by extraction chromatography using a supported liquid anion exchanger: Application to the characterization of high-level nuclear waste solutions. Anal. Chim. Acta 1995, 310, 63–78. [Google Scholar] [CrossRef]
- Liu, B.; Shi, K.L.; Ye, G.Y.; Guo, Z.J.; Wu, W.S. Method development for plutonium analysis in environmental water samples using TEVA microextraction chromatography separation and low background liquid scintillation counter measurement. Microchem. J. 2016, 124, 824–830. [Google Scholar] [CrossRef]
- Maxwell, S.L.; Jones, V.D. Rapid determination of actinides in urine by inductively coupled plasma mass spectrometry and alpha spectrometry: A hybrid approach. Talanta 2009, 80, 143–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maxwell, S.L.; Culligan, B.K.; Jones, V.D.; Nichols, S.T.; Bernard, M.A.; Noyes, G.W. Determination of 237Np and Pu isotopes in large soil samples by inductively coupled plasma mass spectrometry. Anal. Chim. Acta 2010, 682, 130–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.G.; Bu, W.T.; Zheng, J.; Pan, S.M.; Wang, Z.T.; Uchida, S.G. Plutonium determination in seawater by inductively coupled plasma mass spectrometry: A review. Talanta 2016, 151, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, N.; Ishida, J.; Yamato, A.; Iwai, M.; Kinoshita, M. Determination of 239,240Pu and 241Am in Environmental Samples. J. Radioanal. Nucl. Chem. 1987, 115, 369–376. [Google Scholar] [CrossRef]
- GB 14883.8—2016; Determination of Radioactive Substance Plutonium-239 and 240 in Food (Extraction Chromatography). National Health and Family Planning: Beijing, China, 2017.
- Ji, Y.Q. Study on Inductively Coupled Plasma Mass Spectrometry (ICP-MS) Analysis Method of Trace Uranium, Thorium, Neptunium and Plutonium in Environmental Samples. Ph.D. Thesis, China Institute of Atomic Energy, Beijing, China, September 2001. [Google Scholar]
- Ding, W.Z.; Wang, Y.; Huang, H.Q.; Wang, M. Principles of Alpha Spectrum Measurement of Natural Radionuclides. Nucl. Electron. Detect. Technol. 2010, 30, 817–820. [Google Scholar]
- Decay Radiation Search, Decay Radiation Database Version of 9 September, 2021; National Nuclear Data Center (NNDC). Available online: https://www.nndc.bnl.gov/nudat3/indx_dec.jsp (accessed on 30 January 2022).
- Matthews, K.M.; Kim, C.K.; Martin, P. Determination of 210Po in environmental materials: A review of analytical methodology. Appl. Radiat. Isot. 2007, 65, 267–279. [Google Scholar] [CrossRef]
- Solatie, D.; Carbol, P.; Hrnecek, E.; Jaakkola, T.; Betti, M. Sample preparation methods for the determination of plutonium and strontium in environmental samples by low level liquid scintillation counting and α-spectrometry. Radiochim. Acta 2002, 90, 447–454. [Google Scholar] [CrossRef]
- Asgharizadeh, F.; Salimi, B.; Maragheh, M.G.; Mahani, M.K.; Aliabadi, M. Determination of 90Sr concentration in soil and sediment samples from southern shores of Iran using a Sr resin and LSC method. Adv. Liq. Scintill. Spectrom. 2009, 299–303. [Google Scholar]
- Maxwell, S.L.; Culligan, B.K.; Shaw, P.J. Rapid determination of radiostrontium in large soil samples. J. Radioanal. Nucl. Chem. 2013, 295, 965–971. [Google Scholar] [CrossRef] [Green Version]
- Jabbar, T.; Subhani, M.S.; Khan, K.; Rashid, A.; Orfi, S.D.; Khan, A.Y. Natural and fallout radionuclide concentrations in the environment of Islamabad. J. Radioanal. Nucl. Chem. 2003, 258, 143–149. [Google Scholar] [CrossRef]
- Gao, R.Q.; Hou, X.L.; Zhang, L.Y.; Zhang, W.C.; Zhang, M.T. Determination of Ultra-Low Level Plutonium Isotopes in Large Volume Environmental Water Samples. Chin. J. Anal. Chem. 2020, 48, 765–773. [Google Scholar] [CrossRef]
- Varga, B.; Tarjan, S.; Vajda, N. Plutonium isotopes in the Hungarian environment. J. Environ. Radioact. 2008, 99, 641–648. [Google Scholar] [CrossRef]
- Lee, M.H.; Ahn, H.J.; Park, J.H.; Park, Y.J.; Song, K. Rapid sequential determination of Pu, 90Sr and 241Am nuclides in environmental samples using an anion exchange and Sr-Spec resins. Appl. Radiat. Isot. 2011, 69, 295–298. [Google Scholar] [CrossRef]
- Strumińska-Parulska, D.I.; Skwarzec, B.; Fabisiak, J. Plutonium bioaccumulation in seabirds. J. Environ. Radioact. 2011, 102, 1105–1111. [Google Scholar] [CrossRef]
- Arslan, F.; Behrendt, M.; Ernst, W.; Finckh, E.; Greb, G.; Gumbmann, F.; Haller, M.; Hofmann, S.; Karschnick, R.; Klein, M.; et al. 14C and 90Sr measurements at the Erlangen AMS facility. Nucl. Instrum. Methods Phys. Res. B Beam Interact. Mater. At. 1994, 92, 39–42. [Google Scholar] [CrossRef]
- Betti, M.; Giannarelli, S.; Hiernaut, T.; Rasmussen, G.; Koch, L. Detection of trace radioisotopes in soil, sediment and vegetation by glow discharge mass spectrometry. Fresenius’ J. Anal. Chem. 1996, 355, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Wendt, K.; Kratz, J.V.; Lantzsch, J.; Müller, P.; Northershauser, W.; Seibert, A.; Trautmann, N.; Waldek, A.; Zimmer, K. Rapid ultratrace determination of 89Sr and 90Sr in environmental samples by collinear laser resonance ionization spectrometry. Kerntechnik 1997, 62, 81–84. [Google Scholar] [CrossRef]
- Bushaw, B.A.; Cannon, B.D. Diode laser based resonance ionization mass spectrometric measurement of strontium-90. Spectrochim. Acta B At. Spectrosc. 1997, 52, 1839–1854. [Google Scholar] [CrossRef]
- Becker, J.S. Inductively coupled plasma mass spectrometry (ICP-MS) and laser ablation ICP-MS for isotope analysis of long-lived radionuclides. Int. J. Mass Spectrom. 2005, 242, 183–195. [Google Scholar] [CrossRef]
- Alvarez, A.; Navarro, N. Method for actinides and Sr-90 determination in urine samples. Appl. Radiat. Isot. 1996, 47, 869–873. [Google Scholar] [CrossRef]
- Shiraishi, K.; Ko, S.; Arae, H.; Ayama, K. Rapid analysis technique for strontium, thorium, and uranium in urine samples. J. Radioanal. Nucl. Chem. 2007, 273, 307–310. [Google Scholar] [CrossRef]
- Alves, L.; Wiederin, D.; Houk, R. Reduction of Polyatomic Ion Interferences in Inductively Coupled Plasma Mass Spectrometry by Cryogenic Desolvation. Anal. Chem. 1992, 64, 1164–1169. [Google Scholar] [CrossRef] [Green Version]
- Minnich, M.G.; Houk, R.S. Comparison of cryogenic and membrane desolvation for attenuation of oxide, hydride and hydroxide ions and ions containing chlorine in inductively coupled plasma mass spectrometry. J. Anal. At. Spectrom. 1998, 13, 167–174. [Google Scholar] [CrossRef]
- Kim, C.S.; Kim, C.K.; Lee, J.I.; Lee, K.J. Rapid determination of Pu isotopes and atom ratios in small amounts of environmental samples by an on-line sample pre-treatment system and isotope dilution high resolution inductively coupled plasma mass spectrometry. J. Anal. At. Spectrom. 2000, 15, 247–255. [Google Scholar] [CrossRef]
- Ni, Y.; Wang, Z.; Zheng, J.; Tagami, K.; Guo, Q.; Uchida, S.; Tsukada, H. The transfer of fallout plutonium from paddy soil to rice: A field study in Japan. J. Environ. Radioact. 2019, 196, 22–28. [Google Scholar] [CrossRef]
- McCarthy, W.; Nicholls, T.M. Mass-Spectrometric Analysis of Plutonium in Soils Near Sellafield. J. Environ. Radioact. 1990, 12, 1–12. [Google Scholar] [CrossRef]
- Cawse, P.A.; Horrill, A.D. A Survey of Caesium-137 and Plutonium in British Soils in 1977; AERE-R10155; UK Atomic Energy Authority: Harwell, UK, 1986.
- Buesseler, K.O. The isotopic signature of fallout plutonium in the North Pacific. J. Environ. Radioact. 1997, 36, 69–83. [Google Scholar] [CrossRef]
- Chiappini, R.; Pointurier, F.; Millies-Lacroix, J.C.; Lepetit, G.; Hemet, P. 240Pu/239Pu isotopic ratios and 239+240Pu total measurements in surface and deep waters around Mururoa and Fangataufa atolls compared with Rangiroa atoll French Polynesiaž. Sci. Total Environ. 1999, 237–238, 269–276. [Google Scholar] [CrossRef]
- Zhang, W.C.; Lin, J.F.; Fang, S.; Li, C.; Yi, X.; Hou, X.; Chen, N.; Zhang, H.; Xu, Y.; Dang, H.; et al. Determination of ultra-trace level plutonium isotopes in soil samples by triple-quadrupole inductively coupled plasma-mass spectrometry with mass-shift mode combined with UTEVA chromatographic separation. Talanta 2021, 234, 122652. [Google Scholar] [CrossRef]
- Sturup, S.; Dahlgaard, H.; Nielsen, S. High resolution inductively coupled plasma mass spectrometry for the trace determination of plutonium isotopes and isotope ratios in environmental samples. J. Anal. At. Spectrom. 1998, 13, 1321–1326. [Google Scholar] [CrossRef]
- Wang, J.L.; Du, J.Z.; Qu, J.G.; Bi, Q.Q. Distribution of Pu isotopes and 210Pb in the Bohai Sea and Yellow Sea: Implications for provenance and transportation. Chemosphere 2021, 263, 127896. [Google Scholar] [CrossRef]
- Mathew, P.J.; Donovan, A.; David, C.; Michael, A.C.H.; Hirofumi, T.; Kei, O.; Thomas, G.H. Differentiating Fukushima and Nagasaki plutonium from global fallout using 241Pu/239Pu atom ratios: Pu vs. Cs uptake and dose to biota. Sci. Total Environ. 2021, 754, 141890. [Google Scholar]


{kind=link}
| Radionuclide | Weapon Tests | Chernobyl Accident | Fukushima Accident | |||
|---|---|---|---|---|---|---|
| Atmosphere | Ocean | Atmosphere | Ocean | Atmosphere | Ocean | |
| 131I | - | - | 1760 | - | 150–160 | - |
| 137Cs | 950 | 600 | 85 | 16 | 12–20 | 4–27 |
| 90Sr | 600 | 380 | 1 | - | 0.01–0.14 | 0.1–2.2 |
| 239,240Pu | 10.87 | 6.6 | 0.087 | - | (1–2.4) × 10−6 | - |
| 240Pu/239Pu | Ratio | References |
|---|---|---|
| Spent fuel reprocessing | 0.02–0.06 | [13,14] |
| Weapons-grade | <0.06 | [15] |
| Global fallout | 0.176 ± 0.014 | [16] |
| Fukushima | 0.330–0.415 | [17] |
| Chernobyl | 0.4 | [18] |
| Analysis Project | 89Sr, 90Sr | 137Cs | 147Pm | 226Ra | Natural Thorium | Natural Uranium | 239Pu |
|---|---|---|---|---|---|---|---|
| Temperature (°C) | 550 | 450 | 450 | 550 | 550 | 550 | 450 |
| Research Object | Acid Reagent | Digestion Equipment | References |
|---|---|---|---|
| 10 or 20 g soil sample | 1 mL Concentrated HNO3, 150 mL Deionized water | [32] | |
| 5 g plant sample ash | 100 mL 5 mol·L−1 HNO3 | Infrared Lamp | [32] |
| 1 g soil sample | 4.8 mL HF, 1.2 mL HClO4 | Microwave Digestion Apparatus | [33] |
| 50 g reference material + 500 g Qatari soil | 500 mL Concentrated HNO3, 250 mL HCl | Electric heating plate | [34] |
| 0.5 g sludge ash sample from sewage treatment plant | 12 mL HNO3:HCl (3:1) mixture | Microwave Digestion Apparatus | [35] |
| 5 g dried food | 1:1 HNO3, H2O2 | Electric heating plate | [36] |
| 0.5 g food sample | Concentrated HNO3 | Microwave Digestion Apparatus | [37] |
| 1 g soil sample | 4.8 mL HF, 1.2 mL HClO4 | Microwave Digestion Apparatus | [38] |
| Analytical Method | Ray Type | Main Detection Nuclides | Advantage | Disadvantage |
|---|---|---|---|---|
| Alpha spectrometry | α | 238Pu, 239Pu, 210Po, 241Am | Low detection limit and high sensitivity | Complicated process and time-consuming |
| Beta spectrometry | β | 3H, 89Sr, 90Sr, 226Ra, 137Cs | Low background and high detection efficiency | Tedious preprocessing and significant spectrum interference |
| Gamma spectrometry | γ | 55Fe, 60Co, 65Zn, 95Zr, 110Ag, 131I, 134Cs, 137Cs | Simple preprocessing, high detection efficiency, and strong energy resolution | High cost and many influencing factors |
| Liquid scintillation | α, low energy β | 3H, 14C, 89Sr, 90Sr, 239Pu | High detection efficiency, high sensitivity, and high precision | Complicated separation and time-consuming |
| Nuclides | Half-Life | Main Alpha Particles | Main Gamma Rays | Main Beta Particles | |||
|---|---|---|---|---|---|---|---|
| Energy/keV | Intensity/% | Energy/keV | Intensity/% | Energy/keV | Intensity/% | ||
| 90Sr | 28.9 a* | 545.9 | 100.0 | ||||
| 238Pu | 87.7 a | 5499.03 | 70.91 | 43.498 | 0.0392 | ||
| 239Pu | 24,110 a | 5156.59 | 70.77 | 51.624 | 0.0272 | ||
| 240Pu | 6561 a | 5168.17 | 72.8 | 45.244 | 0.0447 | ||
| Sample | ICP–DCR–MS | Radiometric Method | ||
|---|---|---|---|---|
| (pg·g−1) | (Bq·g−1) | (Bq·g−1) 1996 | (Bq·g−1) 2007 | |
| soil 1 | 4.66 ± 0.27 | 23.7 ± 1.3 | 45 ± 9 | 35 ± 7 |
| soil 2 | 13.48 ± 0.68 | 68.6 ± 3.5 | 82 ± 16 | 63 ± 13 |
| soil 3 | 12.9 ± 1.5 | 65.6 ± 7.8 | 99 ± 18 | 76 ± 15 |
| Isotope | ICP–MS/HP/Mistral | Alpha Spectrometry | |
|---|---|---|---|
| Mass/g | Activity/Bq | Activity/Bq | |
| 239Pu | 1.2 × 10−15 | 2.8 × 10−6 | 1 × 10−4 |
| 240Pu | 1.2 × 10−15 | 1.0 × 10−5 | 1 × 10−4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, N.; Li, L.; Yang, X.; Zhao, Y. Analytical Methods for the Determination of 90Sr and 239,240Pu in Environmental Samples. Molecules 2022, 27, 1912. https://doi.org/10.3390/molecules27061912
Zhong N, Li L, Yang X, Zhao Y. Analytical Methods for the Determination of 90Sr and 239,240Pu in Environmental Samples. Molecules. 2022; 27(6):1912. https://doi.org/10.3390/molecules27061912
Chicago/Turabian StyleZhong, Ningjie, Lili Li, Xiaofan Yang, and Yonggang Zhao. 2022. "Analytical Methods for the Determination of 90Sr and 239,240Pu in Environmental Samples" Molecules 27, no. 6: 1912. https://doi.org/10.3390/molecules27061912
APA StyleZhong, N., Li, L., Yang, X., & Zhao, Y. (2022). Analytical Methods for the Determination of 90Sr and 239,240Pu in Environmental Samples. Molecules, 27(6), 1912. https://doi.org/10.3390/molecules27061912