
Computational Methods for Understanding the Selectivity and Signal Transduction Mechanism of Aminomethyl Tetrahydronaphthalene to Opioid Receptors

Abstract
:1. Introduction
2. Results and Discussion
2.1. Chemical Design
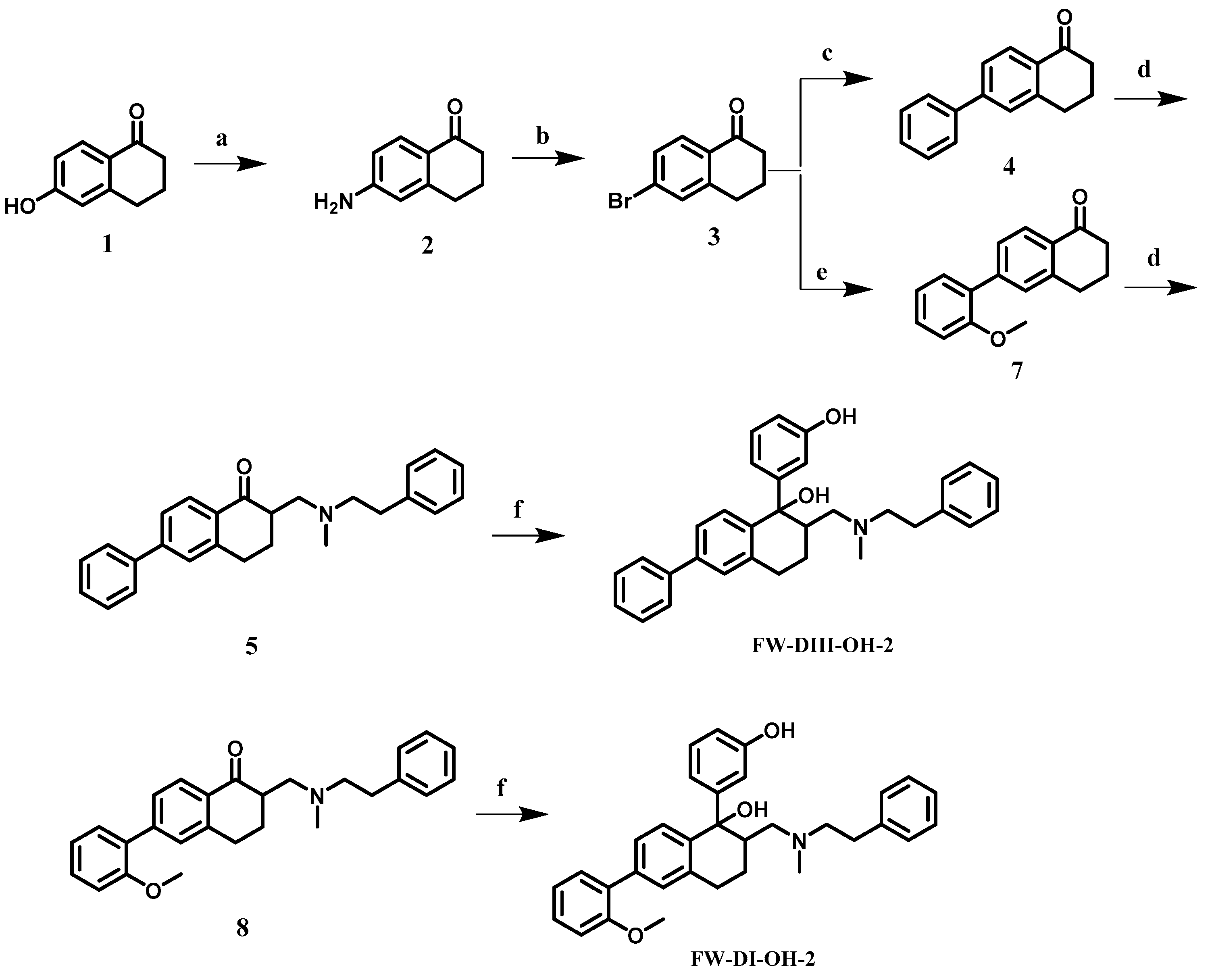
2.2. Chemical Synthesis
2.3. In Vitro Activities
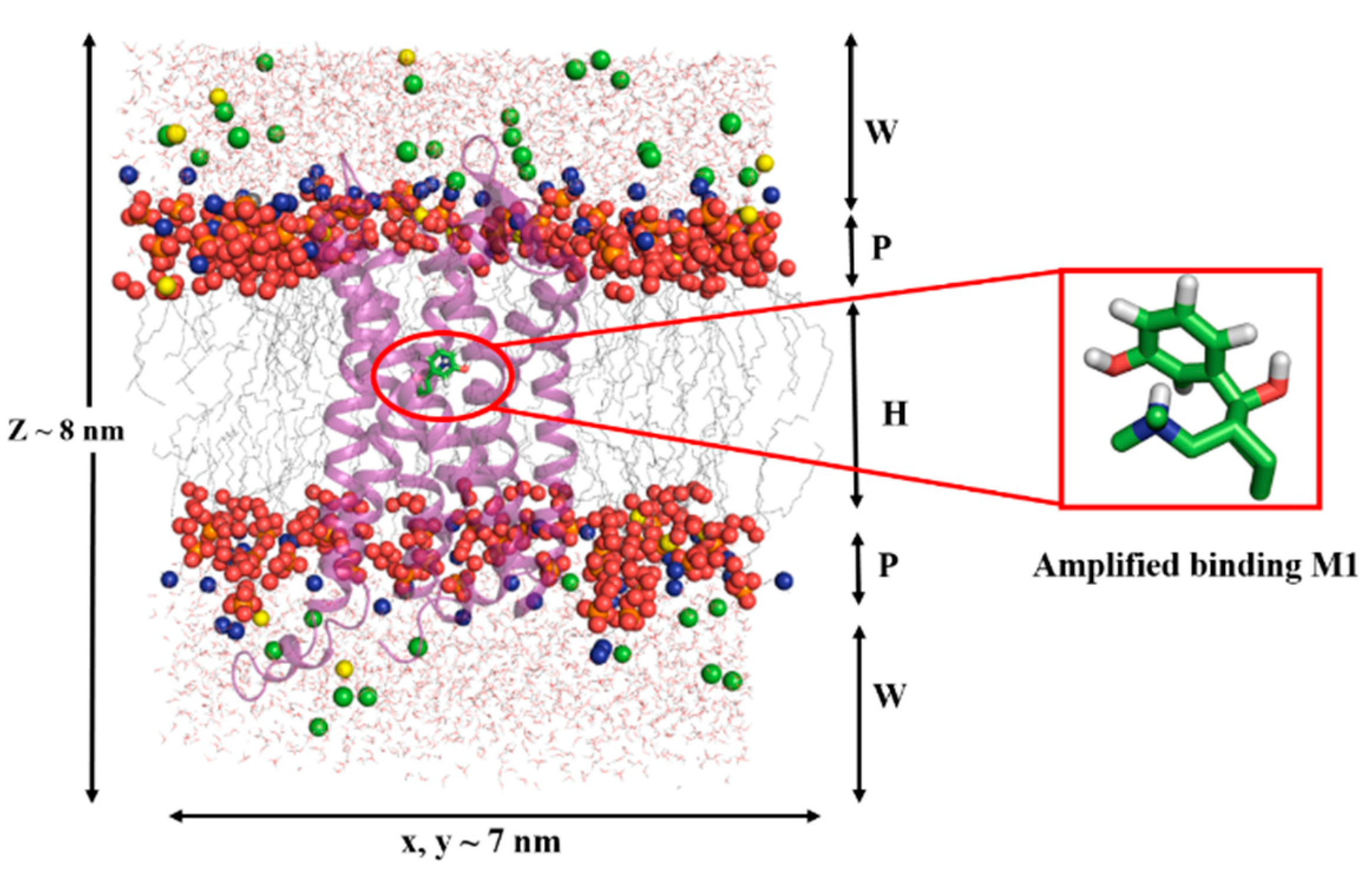
2.4. Results of Molecular Dynamics Simulations
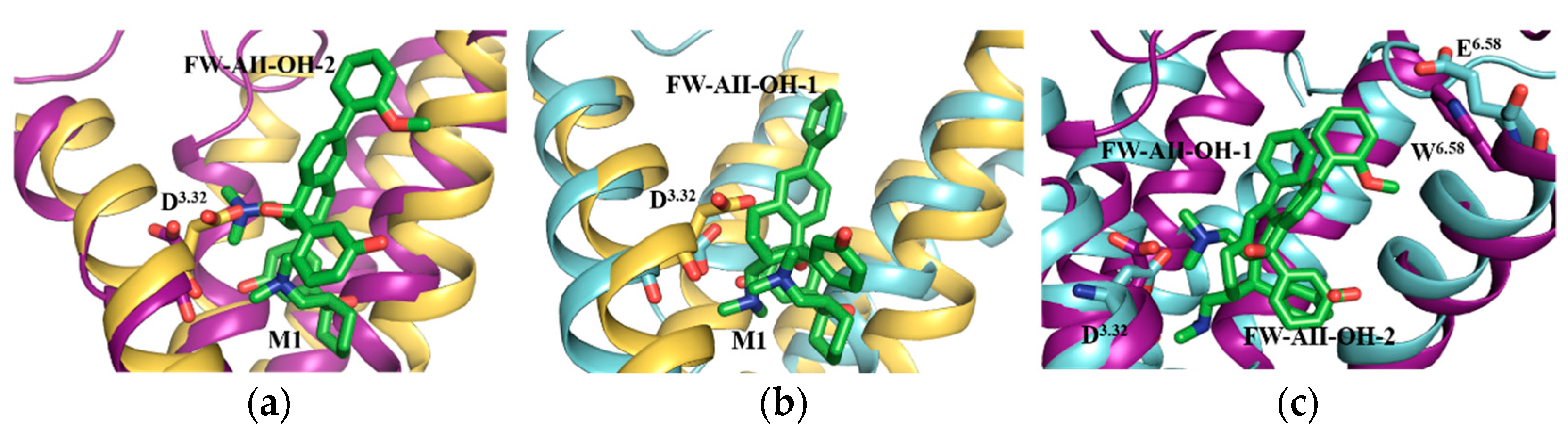
2.5. Selectivity of Probe Molecules
2.6. The Salt Bridges between D3.32 and the Amine Group
2.7. The 3–7 Lock
2.8. The DRY Lock (Connection between TM3 and TM6)
2.9. The Rearrangements of I3.40, P5.50 and F6.44
2.10. The Movements of Transmembrane Helixes
2.11. Transmembrane Helix Movements of Opioid Protein upon Agonist Binding
2.12. The Signal Transduction Mechanisms of MOR, DOR and KOR
3. Materials and Methods
3.1. Chemistry
- 6-amino-3,4-dihydronaphthalen-1(2H)-one, 2
- 6-bromo-3,4-dihydronaphthalen-1(2H)-one 3
- 6-phenyl-3,4-dihydronaphthalen-1(2H)-one 4
- 6-(2-methoxyphenyl)-3,4-dihydronaphthalen-1(2H)-one 7
- General Procedure A for the Preparation of 5 and 8.6-phenyl-2-((methyl(phenethyl)amino)methyl)-3,4-dihydronaphthalen-1(2H)-one, 5
- 6-(2-methoxyphenyl)-2-((methyl(phenethyl)amino)methyl)-3,4-dihydronaphthalen-1(2H)-one, 8
- 1-(3-hydroxyphenyl)-2-((methyl(phenethyl)amino)methyl)-6-phenyl-1,2,3,4-tetrahydronaphthalen-1-ol hydrochloride FW-DIII-OH-2
- 1-(3-hydroxyphenyl)-6-(2-methoxyphenyl)-2-((methyl(phenethyl)amino) methyl)-1,2,3,4-tetrahydronaphthalen-1-ol hydrochloride, FW-DI-OH-2
3.2. Homology Modeling of Human Active MOR
3.3. Protein Preparation
3.4. Molecular Docking
3.5. Molecular Dynamics Simulations
3.6. Cluster Analysis
3.7. Radio Ligand Binding Assay
3.8. [35S]GTP-γ-S Functional Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- ReferencesSchioth, H.B.; Fredriksson, R. The GRAFS classification system of G-protein coupled receptors in comparative perspective. Gen. Comp. Endocrinol. 2005, 142, 94–101. [Google Scholar]
- Waldhoer, M.; Bartlett, S.E.; Whistler, J.L. Opioid receptors. Ann. Rev. Biochem. 2004, 73, 953–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corbett, A.D.; Henderson, G.; McKnight, A.T.; Paterson, S.J. 75 years of opioid research: The exciting but vain quest for the Holy Grail. Br. J. Pharmacol. 2006, 147, S153–S162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kane, B.E.; Svensson, B.; Ferguson, D.M. Molecular recognition of opioid receptor ligands. AAPS J. 2006, 8, E126–E137. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Filizola, M. Opioid receptors: Structural and mechanistic insights into pharmacology and signaling. Eur. J. Pharmacol. 2015, 763 Pt B, 206–213. [Google Scholar] [CrossRef] [Green Version]
- Chou, R.; Fanciullo, G.J.; Fine, P.G.; Adler, J.A.; Ballantyne, J.C.; Davies, P.; Donovan, M.I.; Fishbain, D.A.; Foley, K.M.; Fudin, J.; et al. Clinical guidelines for the use of chronic opioid therapy in chronic noncancer pain. J. Pain 2009, 10, 113–130. [Google Scholar] [CrossRef] [Green Version]
- Soergel, D.G.; Subach, R.A.; Burnham, N.; Lark, M.W.; James, I.E.; Sadler, B.M.; Skobieranda, F.; Violin, J.D.; Webster, L.R. Biased agonism of the mu-opioid receptor by TRV130 increases analgesia and reduces on-target adverse effects versus morphine: A randomized, double-blind, placebo-controlled, crossover study in healthy volunteers. Pain 2014, 155, 1829–1835. [Google Scholar] [CrossRef]
- Manglik, A.; Lin, H.; Aryal, D.K.; McCorvy, J.D.; Dengler, D.; Corder, G.; Levit, A.; Kling, R.C.; Bernat, V.; Hübner, H.; et al. Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. [Google Scholar] [CrossRef] [Green Version]
- Altarifi, A.A.; David, B.; Muchhala, K.H.; Blough, B.E.; Akbarali, H.; Negus, S.S. Effects of acute and repeated treatment with the biased mu-opioid receptor agonist TRV130 (oliceridine) on measures of antinociception, gastrointestinal function, and abuse liability in rodents. J. Psychopharmacol. 2017, 31, 730–739. [Google Scholar] [CrossRef]
- Austin Zamarripa, C.; Edwards, S.R.; Qureshi, H.N.; Yi, J.N.; Blough, B.E.; Freeman, K.B. The G-protein biased mu-opioid agonist, TRV130, produces reinforcing and antinociceptive effects that are comparable to oxycodone in rats. Drug Alcohol. Depend. 2018, 192, 158–162. [Google Scholar] [CrossRef]
- Hill, R.; Disney, A.; Conibear, A.; Sutcliffe, K.; Dewey, W.; Husbands, S.; Bailey, C.; Kelly, E.; Henderson, G. The novel mu-opioid receptor agonist PZM21 depresses respiration and induces tolerance to antinociception. Br. J. Pharmacol. 2018, 175, 2653–2661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kliewer, A.; Schmiedel, F.; Sianati, S.; Bailey, A.; Bateman, J.T.; Levitt, E.S.; Williams, J.T.; Christie, M.J.; Schulz, S. Phosphorylation-deficient G-protein-biased mu-opioid receptors improve analgesia and diminish tolerance but worsen opioid side effects. Nat. Commun. 2019, 10, 367. [Google Scholar] [CrossRef] [PubMed]
- Windh, R.T.; Ajello, C.W.; Leister, L.K.; Gu, M.; Chu, G.; Tuthill, P.A.; Barker, W.M.; Koblish, M.; Wiant, D.D.; Graczyk, T.M.; et al. Potent, orally bioavailable delta opioid receptor agonists for the treatment of pain: Discovery of N,N-diethyl-4-(5-hydroxyspiro[chromene-2,4′-piperidine]-4-yl)benzamide (ADL5859). J. Med. Chem. 2008, 51, 5893–5896. [Google Scholar]
- Le, B.B.; Windh, R.T.; Leister, L.K.; Zhou, Q.; Ajello, C.W.; Chu, G.; Tuthill, P.A.; Barker, W.M.; Koblish, M.; Wiant, D.D.; et al. Spirocyclic Delta Opioid Receptor Agonists for the Treatment of Pain: Discovery of N,N-Diethyl-3-hydroxy-4-(spiro[chromene-2,4′-piperidine]-4-yl) Benzamide (ADL5747). J. Med. Chem. 2009, 52, 5685–5702. [Google Scholar]
- Sorbera, L.A.; Castaner, J.; Leeson, P.A. Nalfurafine Hydrochloride. Drugs Fut. 2003, 28, 237. [Google Scholar] [CrossRef]
- Breslin, H.J.; Diamond, C.J.; Kavash, R.W.; Cai, C.; Dyatkin, A.B.; Miskowski, T.A.; Zhang, S.; Wade, P.R.; Hornby, P.J.; He, W. Identification of a dual δ OR antagonist/μ OR agonist as a potential therapeutic for diarrhea-predominant Irritable Bowel Syndrome (IBS-d). Bioorg. Med. Chem. Lett. 2012, 22, 4869–4872. [Google Scholar] [CrossRef]
- Zheng, Y.; Obeng, S.; Wang, H.; Jali, A.M.; Peddibhotla, B.; Williams, D.A.; Zou, C.; Stevens, D.L.; Dewey, W.L.; Akbarali, H.I.; et al. Design, Synthesis, and Biological Evaluation of the Third Generation 17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6β-[(4′-pyridyl)carboxamido]morphinan (NAP) Derivatives as μ/κ Opioid Receptor Dual Selective Ligands. J. Med. Chem. 2019, 62, 561–574. [Google Scholar] [CrossRef]
- Obeng, S.; Jali, A.; Zheng, Y.; Wang, H.; Schwienteck, K.L.; Chen, C.; Stevens, D.L.; Akbarali, H.I.; Dewey, W.L.; Banks, M.L.; et al. Characterization of 17-Cyclopropylmethyl-3,14β-dihydroxy-4,5α-epoxy-6α-(indole-7-carboxamido)morphinan (NAN) as a Novel Opioid Receptor Modulator for Opioid Use Disorder Treatment. ACS Chem. Neurosci. 2019, 10, 2518–2532. [Google Scholar] [CrossRef]
- Ma, H.; Obeng, S.; Wang, H.; Zheng, Y.; Li, M.; Jali, A.; Stevens, D.L.; Dewey, W.L.; Zhang, Y. Application of Bivalent Bioisostere Concept on Design and Discovery of Potent Opioid Receptor Modulators. J. Med. Chem. 2019, 62, 11399–11415. [Google Scholar] [CrossRef]
- Leppert, W. Tramadol as an analgesic for mild to moderate cancer pain. Pharmacol. Rep. 2009, 61, 978–992. [Google Scholar] [CrossRef]
- Zimmer, O.; Strassburger, W.W.A.; Buschmann, H.H.; Englberger, W.; Friderichs, E.J. Substituted amino Compounds and their use as substances having an analgesic effect. U.S. Patent 6,022,895, 8 February 2000. [Google Scholar]
- Shen, Q.; Qian, Y.; Huang, X.; Xu, X.; Li, W.; Liu, J.; Fu, W. Discovery of Potent and Selective Agonists of δ Opioid Receptor by Revisiting the “Message-Address” Concept. ACS Med. Chem. Lett. 2016, 7, 391–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- Shen, Q.; Qian, Y.; Xu, X.; Li, W.; Liu, J.; Fu, W. Design, synthesis and biological evaluation of N-phenylalkyl-substituted tramadol derivatives as novel μ opioid receptor ligands. Acta Pharmacol. Sin. 2015, 36, 887–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raffa, R.B.; Buschmann, H.; Christoph, T.; Eichenbaum, G.; Englberger, W.; Flores, C.M.; Hertrampf, T.; Kögel, B.; Schiene, K.; Straßburger, W.; et al. Mechanistic and functional differentiation of tapentadol and tramadol. Expert Opin. Pharm. 2012, 13, 1437–1449. [Google Scholar] [CrossRef] [PubMed]
- Claff, T.; Yu, J.; Blais, V.; Patel, N.; Martin, C.; Wu, L.; Han, G.W.; Holleran, B.J.; Van der Poorten, O.; White, K.L.; et al. Elucidating the active δ-opioid receptor crystal structure with peptide and small-molecule agonists. Sci. Adv. 2019, 5, eaax9115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, J.X.; Li, W.; Liu, G.; Zhu, W.; Tang, Y. Computational insights into different inhibition modes of the κ-opioid receptor with antagonists LY2456302 and JDTic. RSC Adv. 2016, 6, 13626–13635. [Google Scholar] [CrossRef]
- Shim, J.; Coop, A.; MacKerell, A.D., Jr. Molecular details of the activation of the mu opioid receptor. J. Phys. Chem. B 2013, 117, 7907–7917. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Palczewski, K.; Peng, Q.; Kolinski, M.; Vogel, H.; Filipek, S. The mechanism of ligand-induced activation or inhibition of mu- and kappa-opioid receptors. Angew. Chem. Int. Ed. Engl. 2015, 54, 7560–7563. [Google Scholar] [CrossRef]
- Huang, W.J.; Manglik, A.; Venkatakrishnan, A.J.; Laeremans, T.; Feinberg, E.N.; Sanborn, A.L.; Kato, H.E.; Livingston, K.E.; Thorsen, T.S.; Kling, R.C.; et al. Structural insights into µ-opioid receptor activation. Nature 2015, 524, 315–321. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Sun, X.; Li, W.; Liu, G.; Tu, Y.; Tang, Y. Molecular switches of the κ opioid receptor triggered by 6′-GNTI and 5′-GNTI. Sci. Rep. 2016, 6, 18913. [Google Scholar] [CrossRef] [Green Version]
- Ballesteros, J.A.; Jensen, A.D.; Liapakis, G.; Rasmussen, S.G.F.; Shi, L.; Gether, U.; Javitch, J.A. Activation of the β2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J. Biol. Chem. 2001, 276, 29171–29177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deupi, X.; Standfuss, J. Structural insights into agonist-induced activation of G-protein-coupled receptors. Curr. Opin. Struct. Biol. 2011, 21, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Marino, K.A.; Yi, S.; Filizola, M. Insights into the function of opioid receptors from molecular dynamics simulations of available crystal structures. Br. J. Pharmacol. 2018, 175, 2834–2845. [Google Scholar] [CrossRef] [PubMed]
- Che, T.; Susruta, M.; Saheem, A.Z.; Pauline, O.; John, D.M.; Wang, S.; Mosier, P.D.; Uprety, R.; Vardy, E.; Krumm, B.E.; et al. Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor. Cell 2018, 172, 55–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Spoel, V.D.; Drunen, R.V. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Comm. 1995, 95, 43–56. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; Spoel, V.D. A package for molecular simulation and trajectory analysis. J. Mol. Mod. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- Aalten, D.M.F.; Bywater, R.; Findlay, J.B.; Hendlich, M.; Hooft, R.W.; Vriend, G. PRODRG, a program for generating molecular topologies and unique molecular descriptors from coordinates of small molecules. J. Comput. Aided Mol. Des. 1996, 10, 255–262. [Google Scholar] [CrossRef]
- Breneman, C.M.; Wiberg, K.B. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- Frisch, M.J. Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2009.
- Lian, P.; Li, L.; Geng, C.; Zhen, X.; Fu, W. Higher-Affinity Agonists of 5-HT1AR Discovered through Tuning the Binding-Site Flexibility. J. Chem. Inf. Model. 2015, 55, 1616–1627. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Shen, J.; Luo, X.; Zhu, W.; Cheng, J.; Yu, K.; Briggs, J.M.; Jin, G.; Chen, K.; Jiang, H. Dopamine D1 receptor agonist and D2 receptor antagonist effects of the natural product (−)-stepholidine: Molecular modeling and dynamics simulations. Biophys. J. 2007, 93, 1431–1441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Li, W.; Du, P.; Yu, K.; Fu, W. Molecular Insights into the D1R Agonist and D2R/D3R Antagonist Effects of the Natural Product (−)-Stepholidine: Molecular Modeling and Dynamics Simulations. J. Phys. Chem. B 2012, 116, 8121–8130. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef] [Green Version]
- Essmannetal, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compds. | Ki (±SEM, nM) or Inhibition (%) | ||
|---|---|---|---|
| MOR a | DOR b | KOR c | |
| M1 [22] | 13.0 ± 0.5 | >104 d | >104 |
| FW-1 | >104 | 21.7 ± 2.9% | 56.3 ± 4.9% |
| FW-AII-OH-1 [22] | 20 ± 1.0% | 22.0 ± 1.0% | 140.0 ± 9.0 nM |
| FW-AII-OH-2 [22] | >104 | 4.7 ± 0.1 nM | 17.7 ± 0.1% |
| FW-DI-OH-2 | NA | 8.65 ± 0.9 nM | 30.1 ± 2.1% |
| FW-DIII-OH-2 | NA | 228.5 ± 5.8 nM | 13.5 ± 0.1% |
| Compounds | MOR | DOR | KOR | |||
|---|---|---|---|---|---|---|
| EC50 (nM) | Emax% | EC50 (nM) | Emax% | EC50 (nM) | Emax% | |
| M1 | 244.7 | 225.7 ± 9 | NA | NA | NA | NA |
| FW-AII-OH-1 [22] | NA | NA | NA | NA | 580 ± 84 | 187 ± 2 |
| FW-AII-OH-2 [22] | NA | NA | 9.3 ± 0.5 | 230 ± 7 | NA | NA |
| DAMGO | 8.06 ± 0.75 | 205.0 ± 2.05 | NA | NA | NA | NA |
| DPDPE | NA | NA | 0.12 ± 0.02 | 216.4 ± 2.20 | NA | NA |
| U50488 | NA | NA | NA | NA | 2.77 ± 0.08 | 199.5 ± 6.30 |
| Complex | Key Residues (Type of Interaction) |
|---|---|
| M1-MOR | Y7.43 (π-π interaction) |
| FW-AII-OH-1-KOR | Y7.43 (π-π interaction), I6.55 and I7.39 (hydrophobic interaction) |
| FW-AII-OH-2-DOR | W6.58 (π-π interaction), V6.55, F6.54, I7.39 and W6.48 (hydrophobic interaction) |
| FW-DI-OH-2-DOR FW-DIII-OH-2-DOR | Y7.43 and W6.58 (π-π interaction), V6.55, F6.54, I7.39 and W6.48 (hydrophobic interaction) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, P.; Zhang, J.; Chen, B.; Li, X.; Zhang, W.; Zhu, M.; Li, W.; Li, J.; Fu, W. Computational Methods for Understanding the Selectivity and Signal Transduction Mechanism of Aminomethyl Tetrahydronaphthalene to Opioid Receptors. Molecules 2022, 27, 2173. https://doi.org/10.3390/molecules27072173
Xie P, Zhang J, Chen B, Li X, Zhang W, Zhu M, Li W, Li J, Fu W. Computational Methods for Understanding the Selectivity and Signal Transduction Mechanism of Aminomethyl Tetrahydronaphthalene to Opioid Receptors. Molecules. 2022; 27(7):2173. https://doi.org/10.3390/molecules27072173
Chicago/Turabian StyleXie, Peng, Junjie Zhang, Baiyu Chen, Xinwei Li, Wenbo Zhang, Mengdan Zhu, Wei Li, Jianqi Li, and Wei Fu. 2022. "Computational Methods for Understanding the Selectivity and Signal Transduction Mechanism of Aminomethyl Tetrahydronaphthalene to Opioid Receptors" Molecules 27, no. 7: 2173. https://doi.org/10.3390/molecules27072173
APA StyleXie, P., Zhang, J., Chen, B., Li, X., Zhang, W., Zhu, M., Li, W., Li, J., & Fu, W. (2022). Computational Methods for Understanding the Selectivity and Signal Transduction Mechanism of Aminomethyl Tetrahydronaphthalene to Opioid Receptors. Molecules, 27(7), 2173. https://doi.org/10.3390/molecules27072173