
Synthesis and Hemostatic Activity of New Amide Derivatives

 , , , , ,
, , , , ,
Abstract
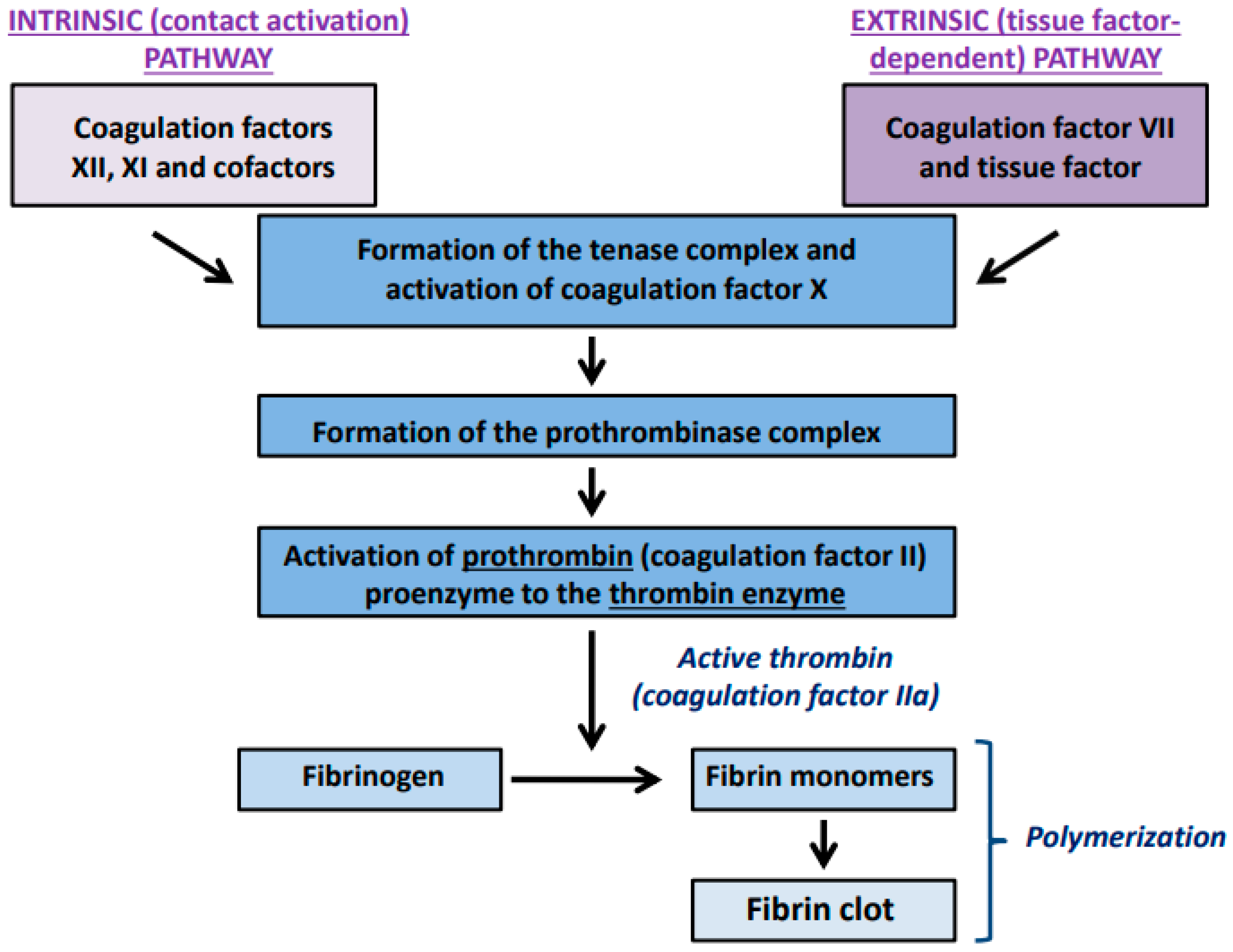
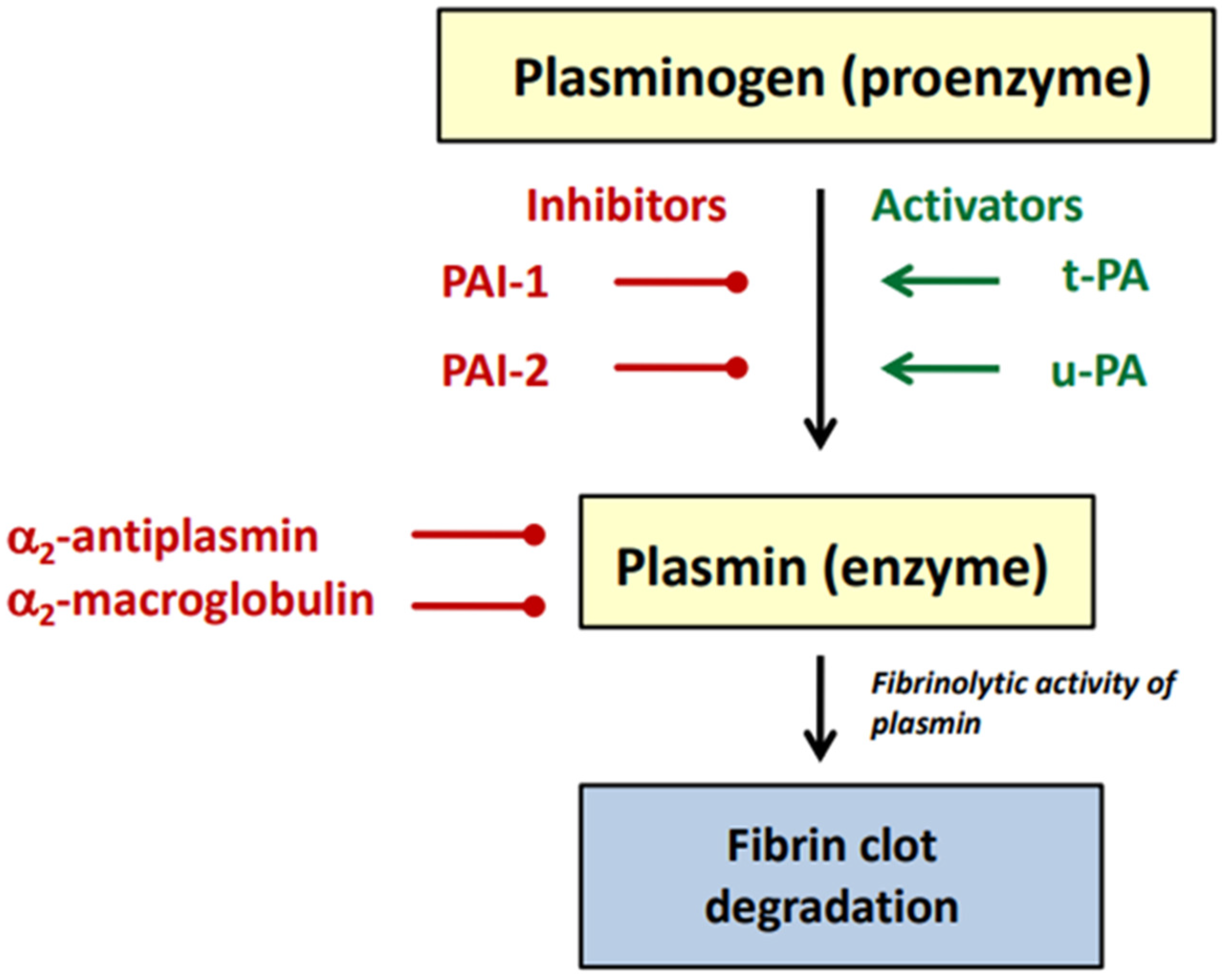
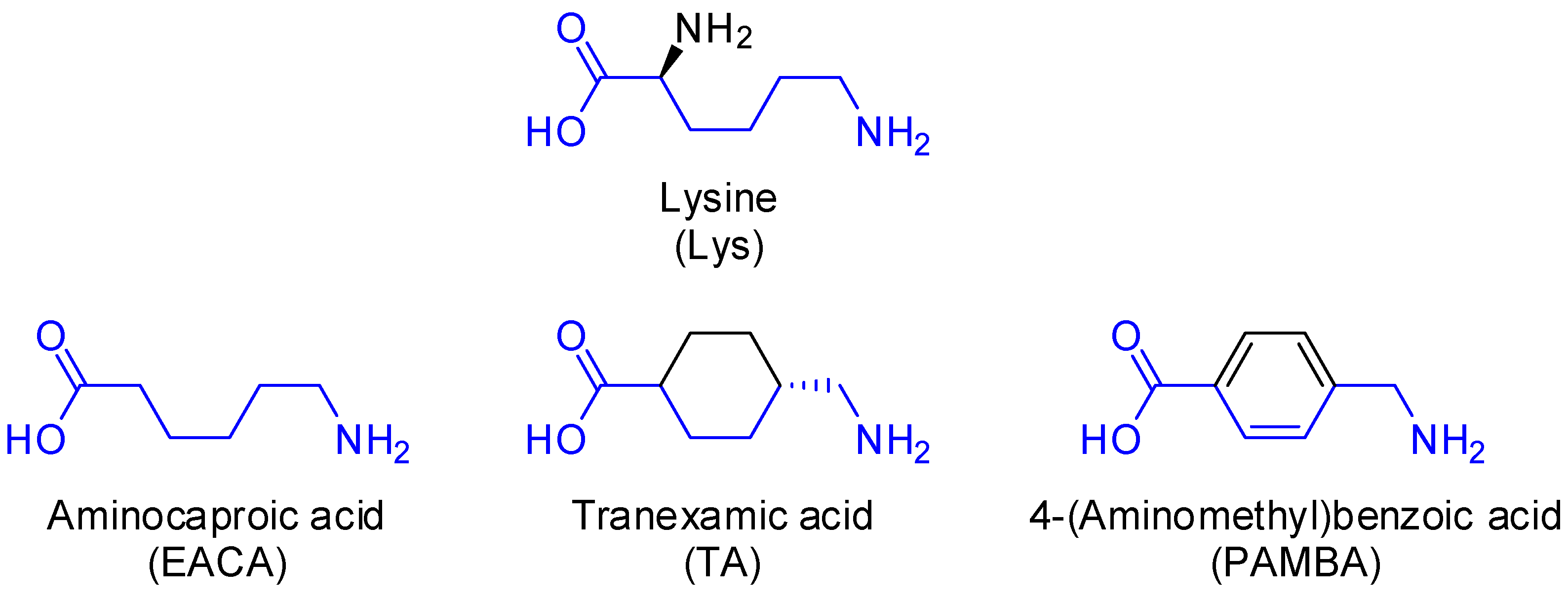
:1. Introduction
2. Results and Discussion
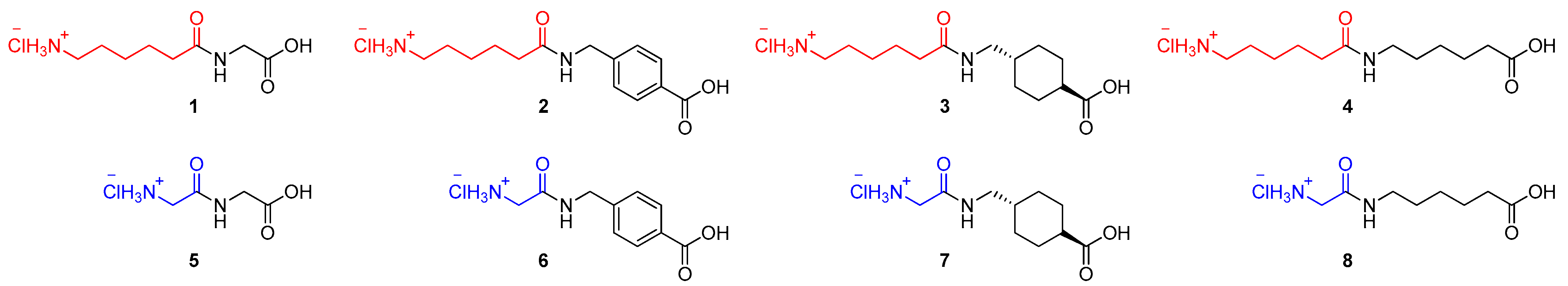
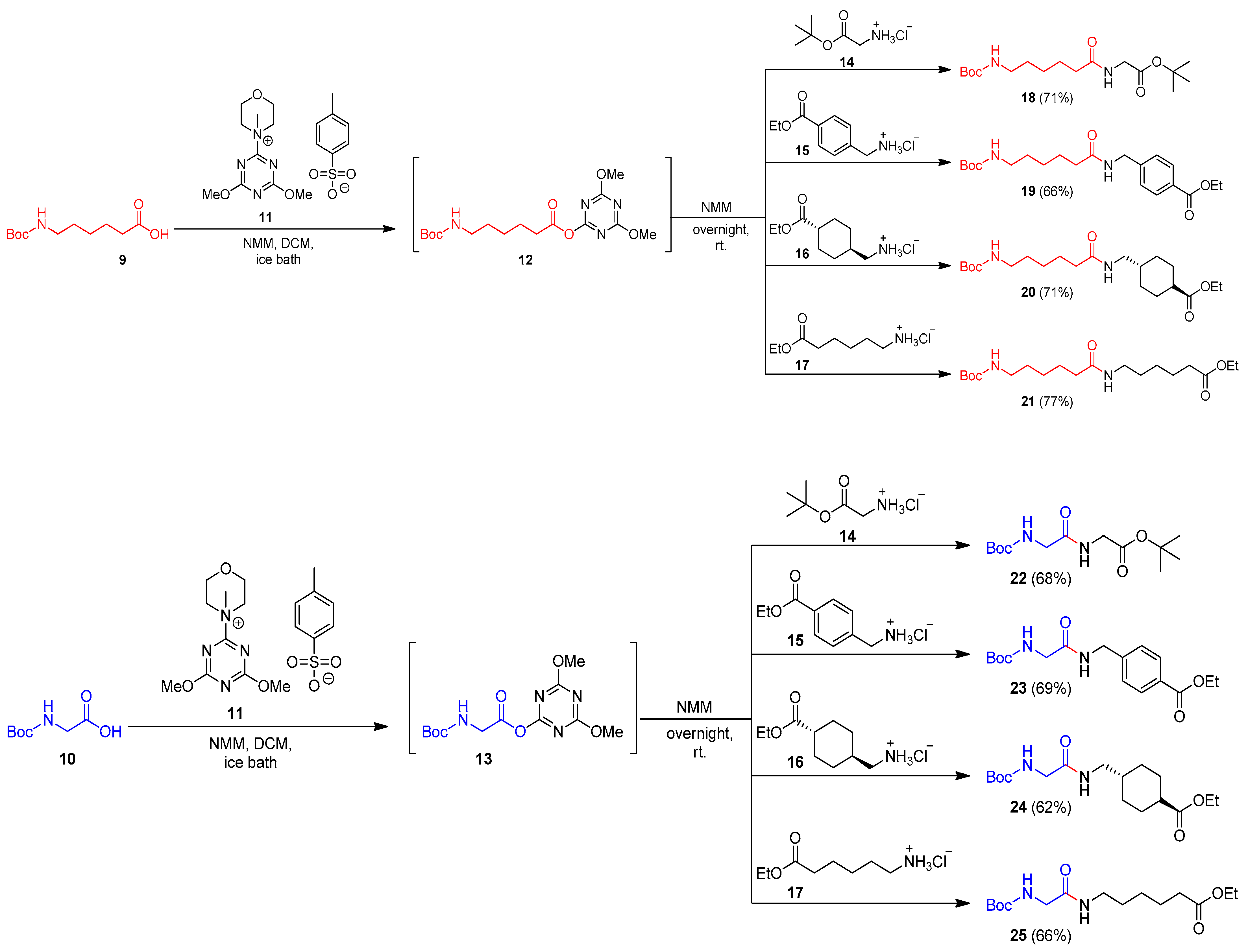
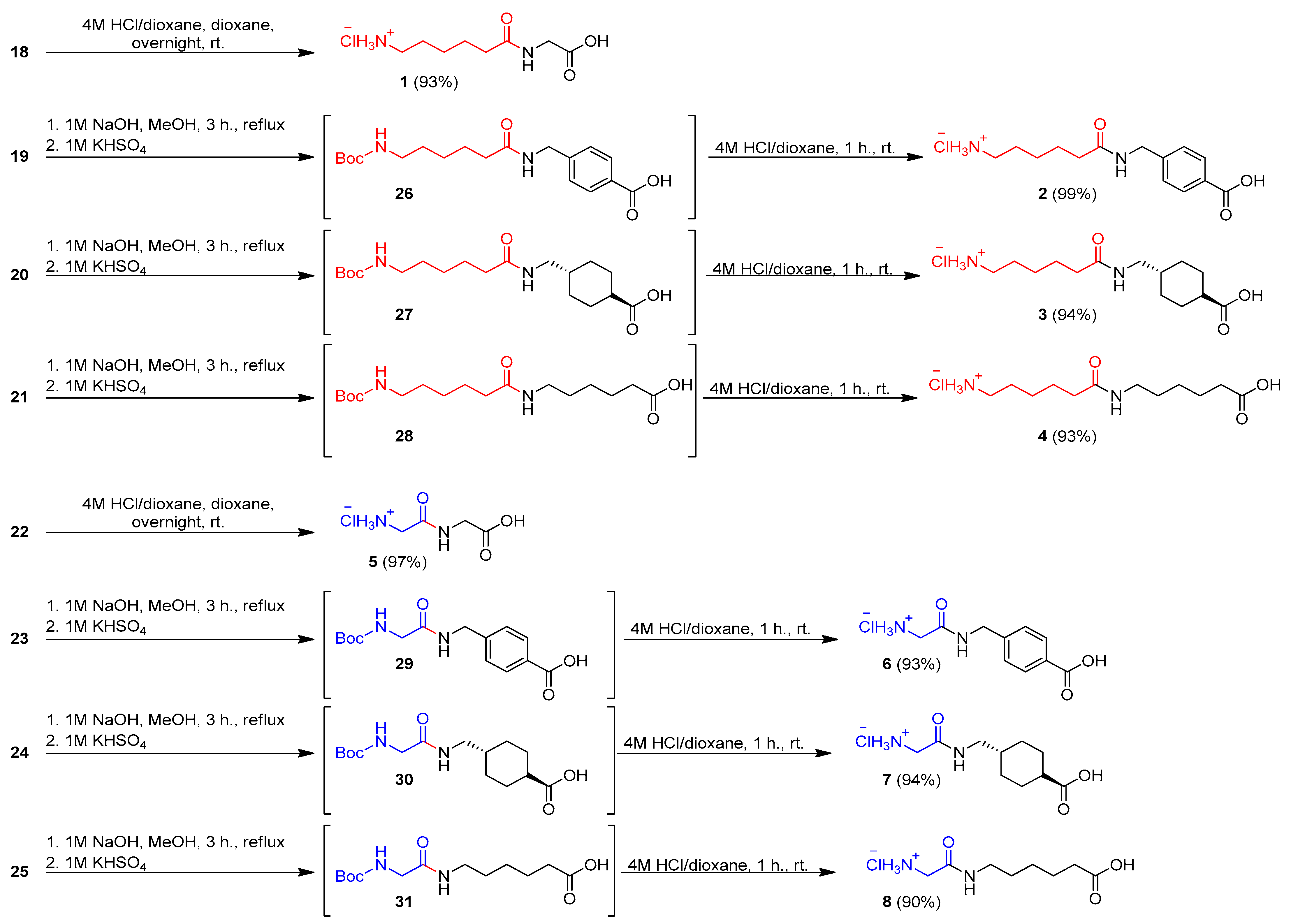
2.1. Chemistry
2.2. Biological Activity
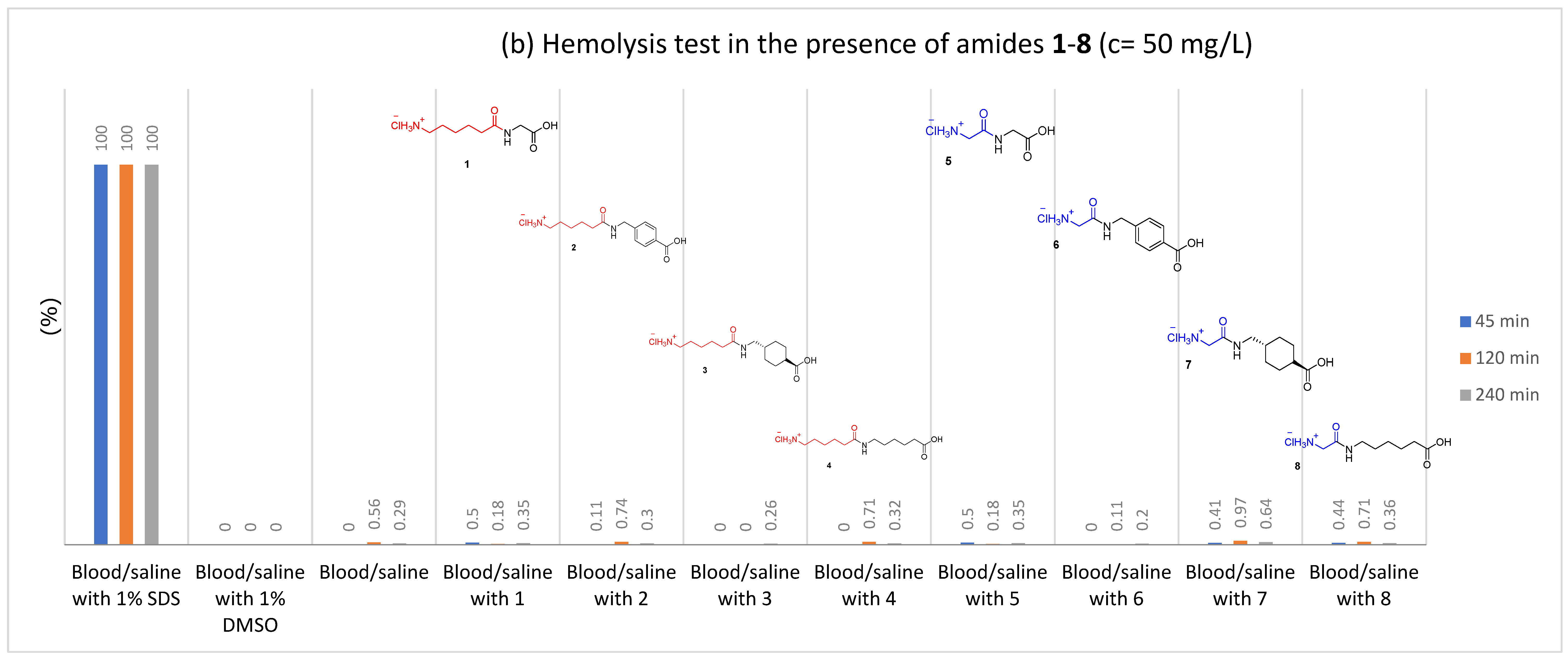
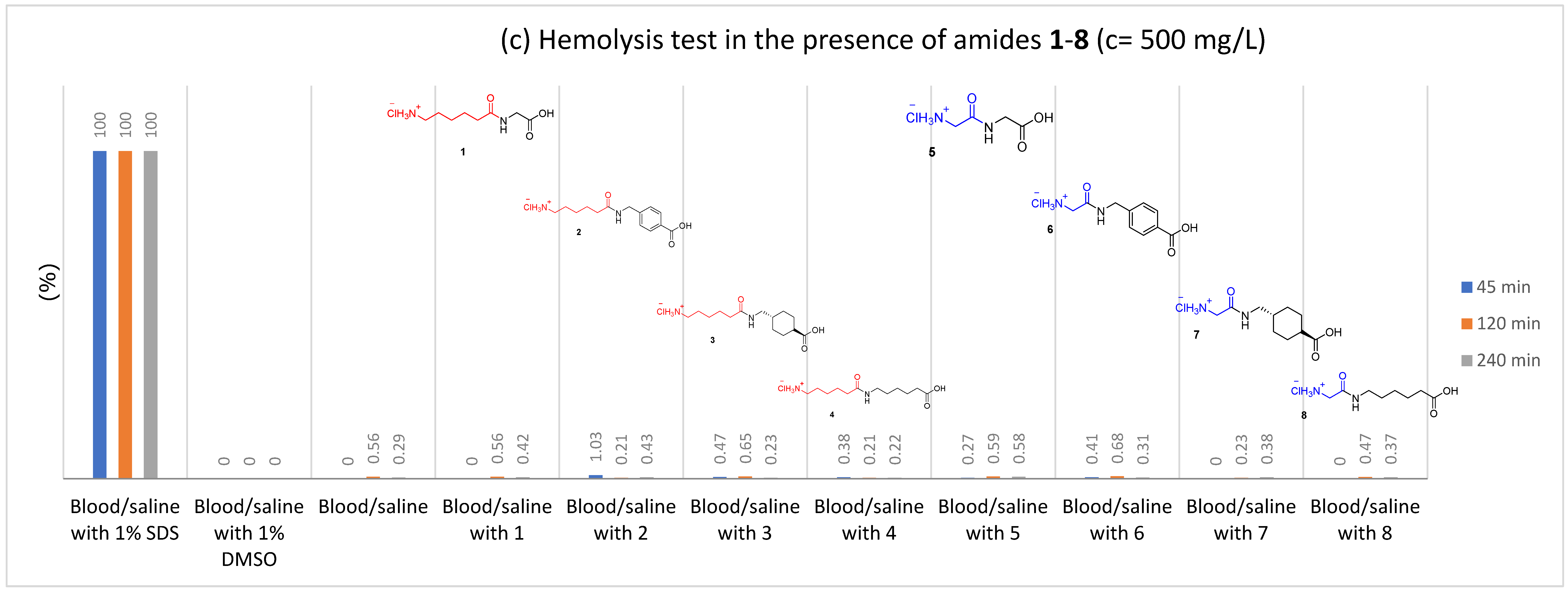
2.2.1. Hemolysis
2.2.2. Clotting Assays and Determination of Fibrinolytic Efficiency
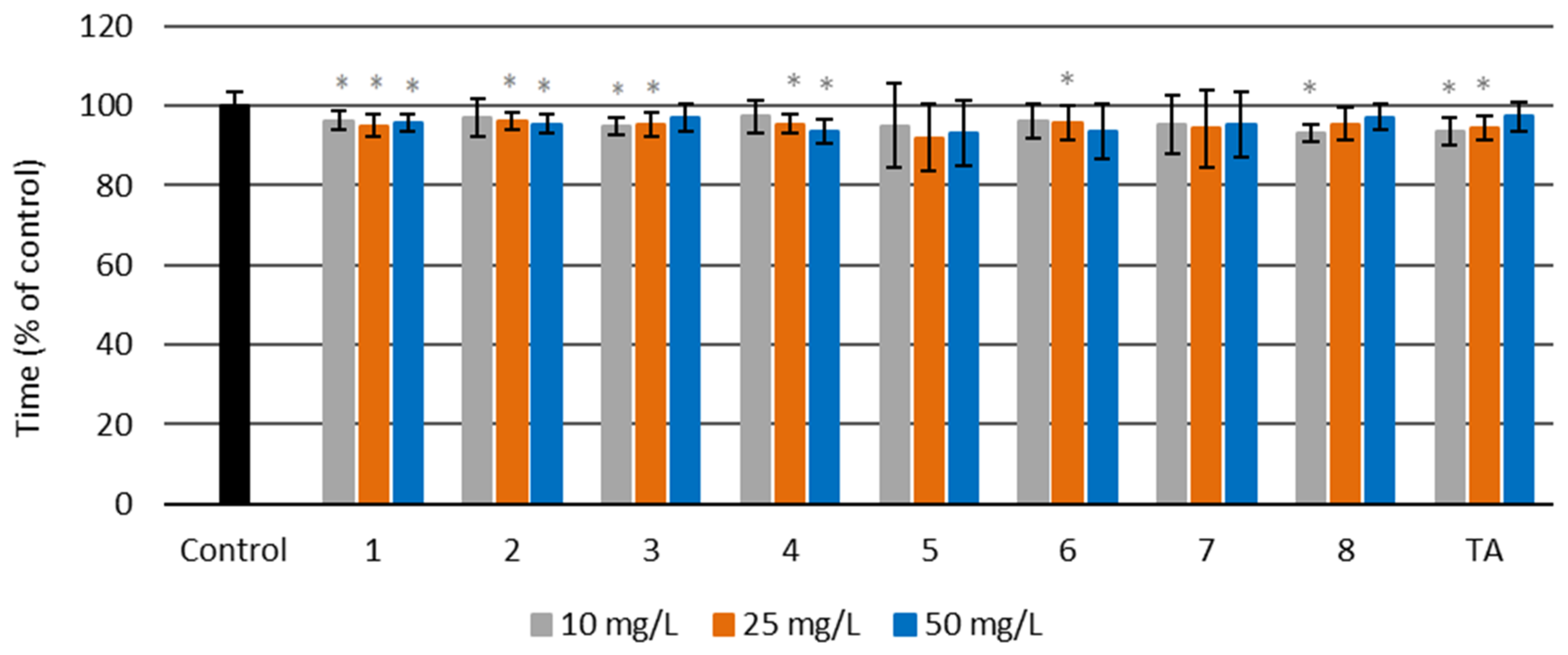
Prothrombin Time (PT)
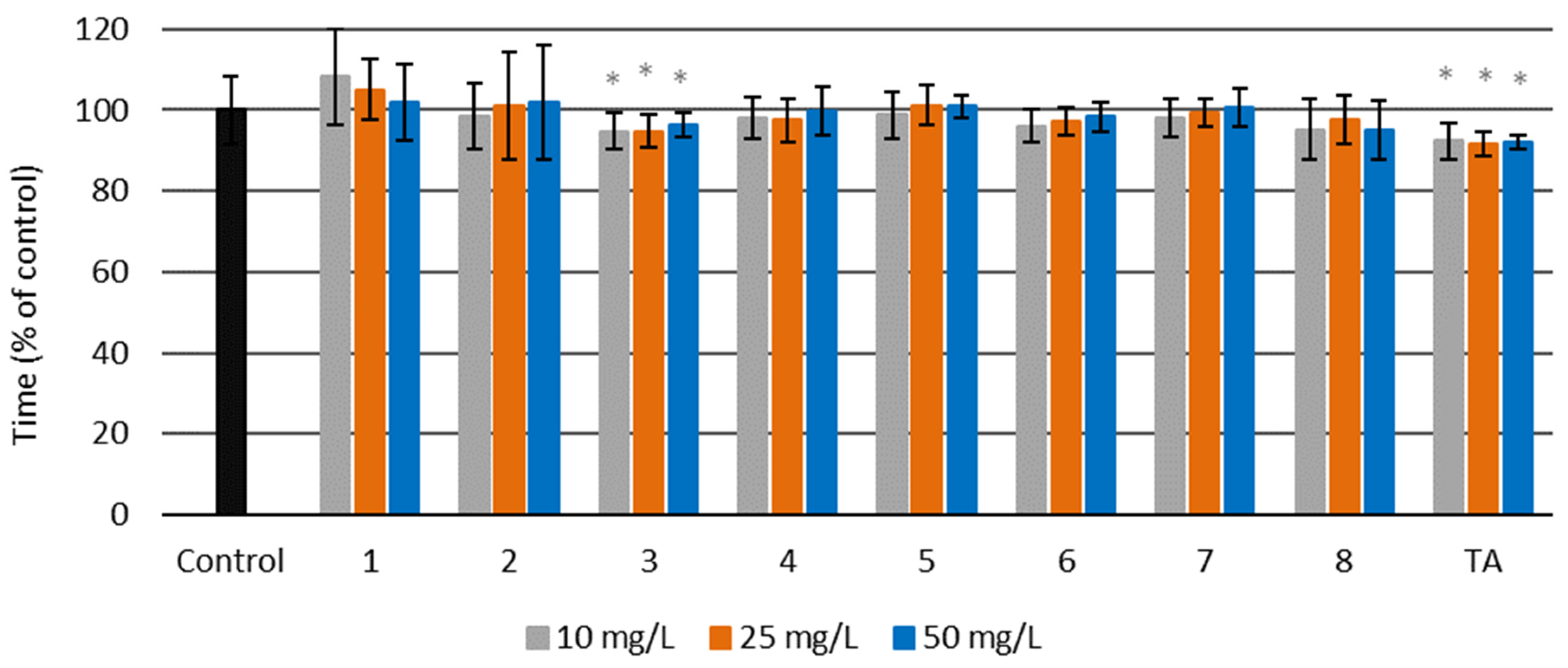
Activated Partial Thromboplastin Time (aPTT)
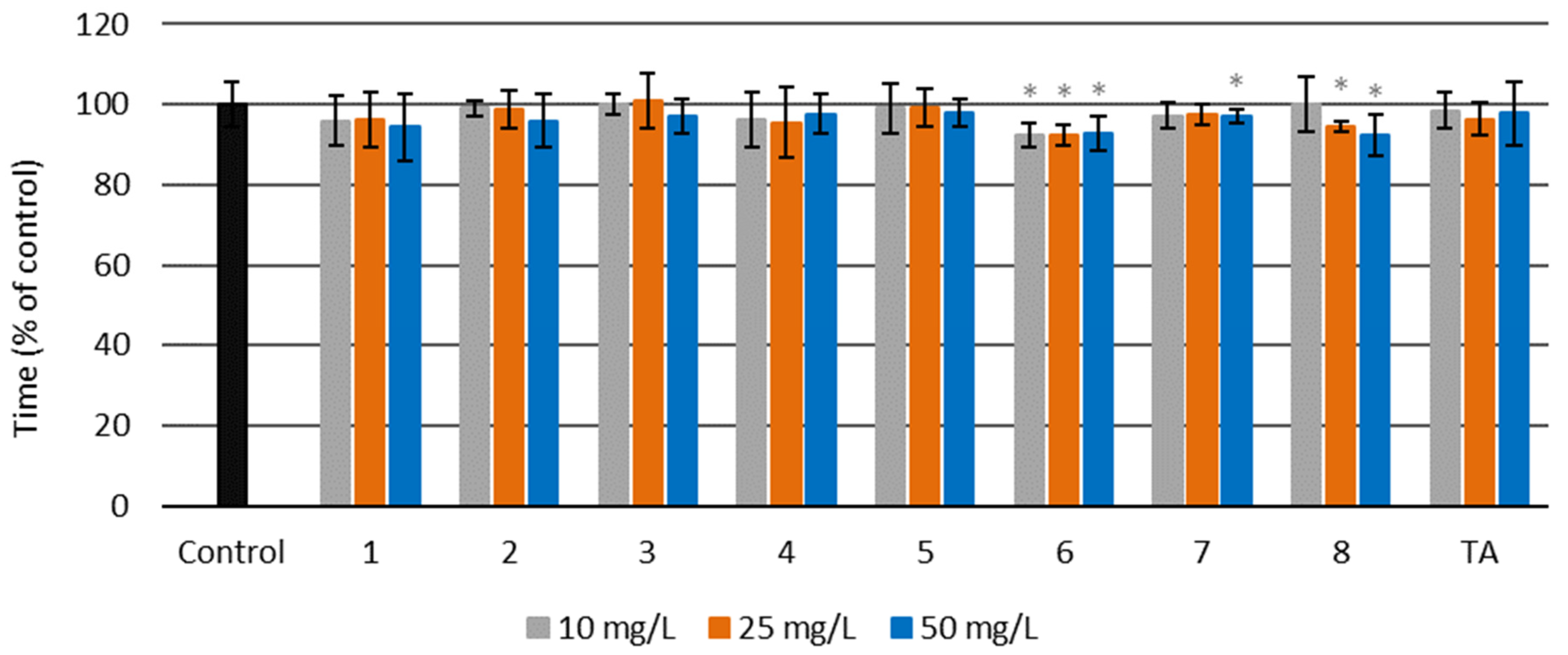
Thrombin Time (TT)
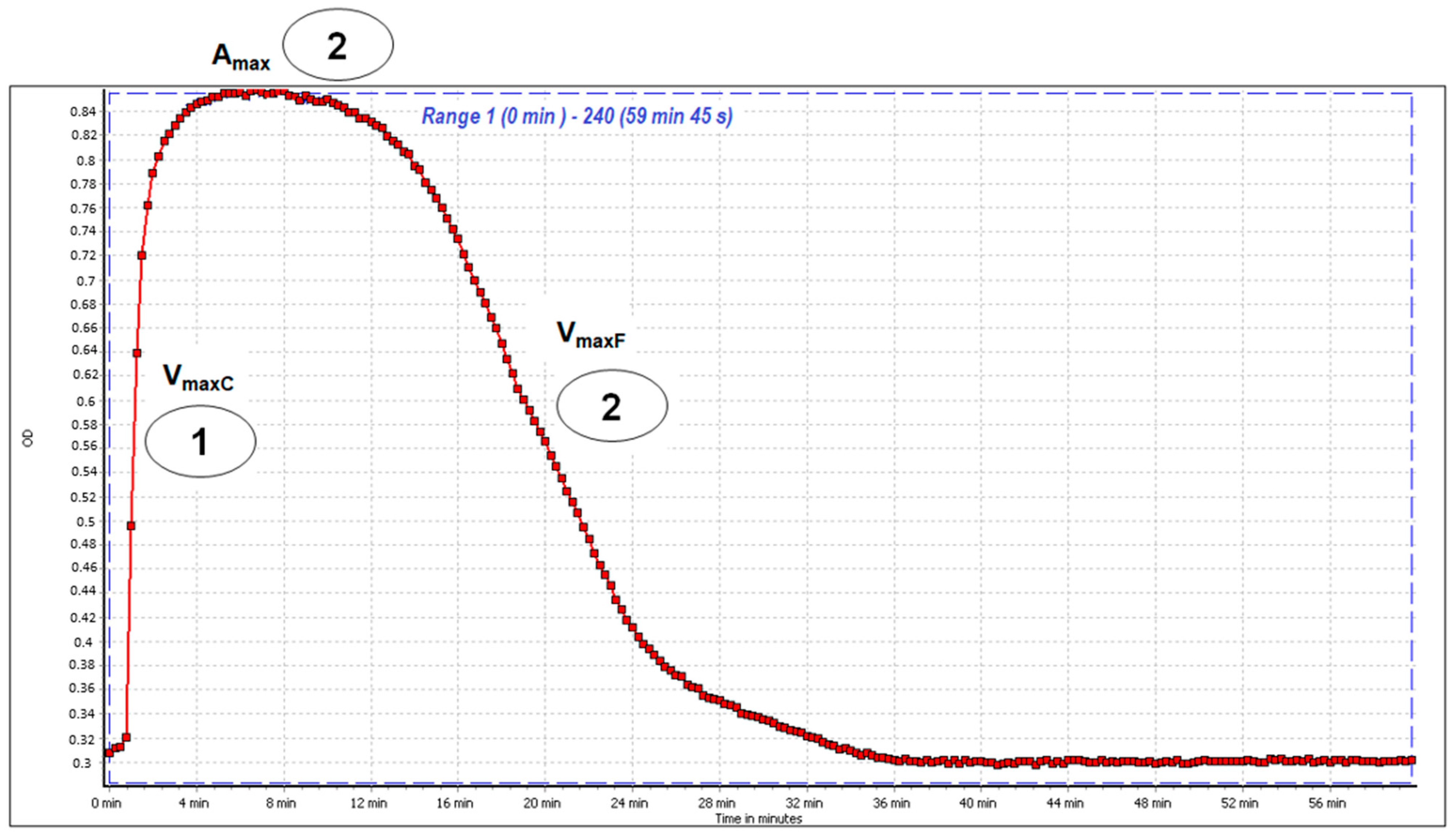
Clot Formation and Fibrinolysis (CFF) Assay
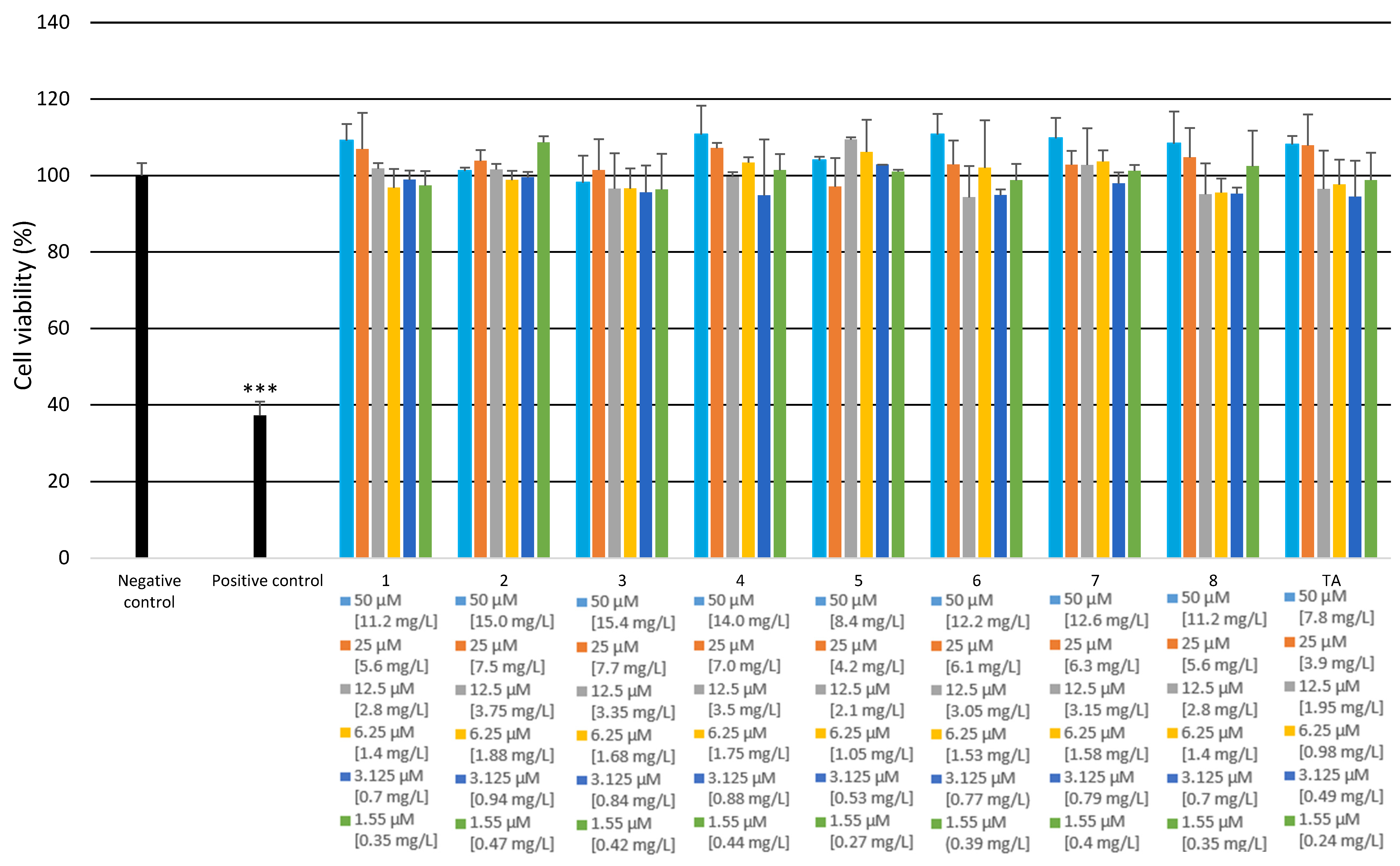
2.2.3. Cytotoxicity and Genotoxicity Analysis
3. Materials and Methods
3.1. General Procedures for the Synthesis of N-Boc Amides 18–25
3.2. General Procedure for Synthesis Hydrochlorides of Amides 1 and 5
3.3. General Procedure for Synthesis Hydrochlorides of Amides 2–4 and 5–7
3.4. Evaluation of the Biological Activity of the Synthesized Compounds
3.4.1. Hemolysis Assay
3.4.2. Blood Plasma Clotting Assays
3.4.3. Prothrombin Time (PT)
3.4.4. Activated Partial Thromboplastin Time (aPTT)
3.4.5. Thrombin Time (TT)
3.4.6. Clot Formation and Fibrinolysis (CFF) Assay
3.4.7. Cell Culture
3.4.8. Cytotoxicity Analysis
3.4.9. Genotoxicity Analysis
3.4.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- McCormack, P.L. Tranexamic acid. A review of its use in the treatment of hyperfibrinolysis. Drugs 2012, 72, 585–617. [Google Scholar] [CrossRef]
- Mahdy, A.M.; Webster, N.R. Perioperative systemic hemostatic agents. Br. J. Anaesth. 2004, 93, 842–858. [Google Scholar] [CrossRef] [Green Version]
- Dunn, C.J.; Goa, K.L. Tranexamic acid: A review of its use in surgery and other indications. Drugs 1999, 57, 1005–1032. [Google Scholar] [CrossRef] [PubMed]
- Wellington, K.; Wagstaff, A.J. Tranexamic acid. A review of its use in the management of menorrhagia. Drugs 2003, 63, 1417–1433. [Google Scholar] [CrossRef]
- Pharmacia & Upjohn. Cyklokapron. ABPI Compendium of Data Sheets and Summaries of Product Characteristics 1998–1999; Datapharm Publications Ltd.: London, UK, 2010. [Google Scholar]
- Pilbrant, A.; Schannong, M.; Vessman, J. Pharmacokinetics and bioavailability of tranexamic acid. Eur. J. Clin. Pharmacol. 1981, 20, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Andersson, L.; Nilsson, I.M.; Niléhn, J.E.; Hedner, U.; Granstrand, B.; Melander, B. Experimental and clinical studies on AMCA, the antifibrinolytically active isomer of p-aminomethyl cyclohexane carboxylic acid. Scand. J. Haematol. 1965, 2, 230–247. [Google Scholar] [CrossRef] [PubMed]
- Widlund, L.; Strömberg, S.; Hellström, H.; Osanius, B. The Disposition of Tranexamic Acid (AMCA) in Various Animal Species and in Man after Oral Dosage; Kabi AB: Stockholm, Sweden, 1979. [Google Scholar]
- Pfizer. Cyklokapron_ (Tranexamic Acid Injection): US Prescribing Information. Available online: http://labeling.pfizer.com/ShowLabeling.aspx?id=556 (accessed on 3 April 2021).
- Daiichi Pharmaceutical Co., Ltd. Transamin_ (Tranexamic Acid) Injection 5%, 10%: Japanese Prescribing Information; Daiichi Pharmaceutical Co., Ltd.: Tokyo, Japan, 2005. [Google Scholar]
- Electronic Medicines Compendium. Cyklokapron Tablets: Summary of Product Characteristics. Available online: https://www.medicines.org.uk/emc/product/100/smpc#gref (accessed on 14 April 2021).
- Brown, R.S.; Thwaites, B.K.; Mongan, P.D. Tranexamic acid is effective in decreasing postoperative bleeding and transfusions in primary coronary artery bypass operations: A double-blind, randomized, placebo-controlled trial. Anesth. Analg. 1997, 85, 963–970. [Google Scholar] [CrossRef]
- Coffey, A.; Pittmam, J.; Halbrook, H.; Fehrenbacher, J.; Beckman, D.; Hormuth, D. The use of tranexamic acid to reduce postoperative bleeding following cardiac surgery: A double-blind randomized trial. Am. Surg. 1995, 61, 566–568. [Google Scholar] [PubMed]
- Horrow, J.C.; Van Riper, D.F.; Strong, M.D.; Grunewald, K.E.; Parmet, J.L. The dose-response relationship of tranexamic acid. Anesthesiology 1995, 82, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Menichetti, A.; Tritapepe, L.; Ruvolom, G.; Speziale, G.; Cogliati, A.; Di Giovanni, C.; Pacilli, M.; Criniti, A. Changes in coagulation patterns, blood loss and blood use after cardiopulmonary bypass: Aprotinin vs tranexamic acid vs epsilon aminocaproic acid. J. Cardiovasc. Surg. 1996, 37, 401–407. [Google Scholar]
- Pugh, S.C.; Wielogorski, A.K. A comparison of the effects of tranexamic acid and low-dose aprotinin on blood loss and homologous blood usage in patients undergoing cardiac surgery. J. Cardiothorac. Vasc. Anesth. 1995, 9, 240–244. [Google Scholar] [CrossRef]
- Speekenbrink, R.G.; Vonk, A.B.; Wildevuur, C.R.; Eijsman, L. Hemostatic efficacy of dipyridamole, tranexamic acid, and aprotinin in coronary bypass grafting. Ann. Thorac. Surg. 1995, 59, 438–442. [Google Scholar] [CrossRef]
- Casati, V.; Guzzon, D.; Oppizzi, M.; Bellotti, F.; Franco, A.; Gerli, C.; Cossolini, M.; Torri, G.; Calori, G.; Benussi, S.; et al. Tranexamic acid compared with high-dose aprotinin in primary elective heart operations: Effects on perioperative bleeding and allogeneic transfusions. J. Thorac. Cardiovasc. Surg. 2000, 120, 520–527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, W.; Spannagl, M.; Boehm, J.; Hauner, K.; Braun, S.; Schuster, T.; Busley, R. Tranexamic acid and aprotinin in primary cardiac operations: An analysis of 220 cardiac surgical patients treated with tranexamic acid or aprotinin. Anesth. Analg. 2008, 107, 1469–1478. [Google Scholar] [CrossRef] [PubMed]
- Benoni, G.; Fredin, H. Fibrinolytic inhibition with tranexamic acid reduces blood loss and blood transfusion after knee arthroplasty: A prospective, randomised, double-blind study of 86 patients. J. Bone Jt. Surg. Br. 1996, 78, 434–440. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.; Abrishami, A.; El Beheiry, H.; Mahomed, N.N.; Davey, J.R.; Gandhi, R.; Syed, K.A.; Hasan, S.M.O.; De Silva, Y.; Chung, F. Topical application of tranexamic acid reduces postoperative blood loss in total knee arthroplasty: A randomized, controlled trial. J. Bone Jt. Surg. 2010, 92, 2503–2513. [Google Scholar] [CrossRef]
- Johansson, T.; Pettersson, L.G.; Lisander, B. Tranexamic acid in total hip arthroplasty saves blood and money: A randomized, double-blind study in 100 patients. Acta Orthop. 2005, 76, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Elwatidy, S.; Jamjoom, Z.; Elgamal, E.; Zakaria, A.; Turkistani, A.; El-Dawlatly, A. Efficacy and safety of prophylactic large dose of tranexamic acid in spine surgery: A prospective, randomized, double-blind, placebo-controlled study. Spine 2008, 33, 2577–2580. [Google Scholar] [CrossRef]
- Dalmau, A.; Sabate, A.; Koo, M.; Bartolome, C.; Rafecas, A.; Figueras, J.; Jaurrieta, E. The prophylactic use of tranexamic acid and aprotinin in orthotopic liver transplantation: A comparative study. Liver Transplant. 2004, 10, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Rannikko, A.; Pe’tas, A.; Taari, K. Tranexamic acid in control of primary hemorrhage during transurethral prostatectomy. Urology 2004, 64, 955–958. [Google Scholar] [CrossRef] [PubMed]
- Bonnar, J.; Sheppard, B.L. Treatment of menorrhagia during menstruation: Randomised controlled trial of ethamsylate, mefenamic acid, and tranexamic acid. BMJ 1996, 313, 579–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukes, A.S.; Moore, K.A.; Muse, K.N.; Gersten, J.K.; Hecht, B.R.; Edlund, M.; Richter, H.E.; Eder, S.E.; Attia, G.R.; Patrick, D.L.; et al. Tranexamic acid treatment for heavy menstrual bleeding: A randomized controlled trial. Obstet. Gynecol. 2010, 116, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Gai, M.-Y.; Wu, L.-F.; Su, Q.-F.; Tatsumoto, K. Clinical observation of blood loss reduced by tranexamic acid during and after caesarian section: A multi-center, randomized trial. Eur. J. Obstet. Gynecol. Reprod. Biol. 2004, 112, 154–157. [Google Scholar] [CrossRef]
- Movafegh, A.; Eslamian, L.; Dorabadi, A. Effect of intravenous tranexamic acid administration on blood loss during and after cesarean delivery. Int. J. Gynaecol. Obstet. 2011, 115, 224–226. [Google Scholar] [CrossRef]
- Barer, D.; Ogilvie, A.; Henry, D.; Dronfield, M.; Coggon, D.; French, S.; Ellis, S.; Atkinson, M.; Langman, M. Cimetidine and tranexamic acid in the treatment of acute upper-gastrointestinal-tract bleeding. N. Engl. J. Med. 1983, 308, 1571–1575. [Google Scholar] [CrossRef]
- Biggs, J.C.; Hugh, T.B.; Dodds, A.J. Tranexamic acid and upper gastrointestinal haemorrhag: A double-blind trial. Gut 1976, 17, 729–734. [Google Scholar] [CrossRef]
- Borea, G.; Montebugnoli, L.; Capuzzi, P.; Magelli, C. Tranexamic acid as a mouthwash in anticoagulant-treated patients undergoing oral surgery: An alternative method to discontinuing anticoagulant therapy. Oral Surg. Oral Med. Oral Pathol. 1993, 75, 29–31. [Google Scholar] [CrossRef]
- Porte, R.J.; Leebeek, F.W. Pharmacological strategies to decrease transfusion requirements in patients undergoing surgery. Drugs 2002, 62, 2193–2211. [Google Scholar] [CrossRef]
- Levi, M.; Cromheecke, M.E.; de Jonge, E.; Prins, M.H.; de Mol, B.J.; Briet, E.; Buller, H.R. Pharmacological strategies to decrease excessive blood loss in cardiac surgery: A meta-analysis of clinically relevant endpoints. Lancet 1999, 354, 1940–1947. [Google Scholar] [CrossRef]
- Munoz, J.J.; Birkmeyer, N.J.; Birkmeyer, J.D.; O’Connor, G.T.; Dacey, L.J. Is epsilon-aminocaproic acid as effective as aprotinin in reducing bleeding with cardiac surgery?: A meta-analysis. Circulation 1999, 99, 81–89. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.; Lewis, J.H.; Navalgund, A.; Russell, M.W.; Bontempo, F.A.; Niren, L.S.; Starzl, T.E. Epsilon-aminocaproic acid for treatment of fibrinolysis during liver transplantation. Anesthesiology 1987, 66, 766–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franck, M.; Sladen, R.N. Drugs to prevent and reverse anticoagulation. Anesthesiol. Clin. N. Am. 1999, 17, 799–811. [Google Scholar] [CrossRef]
- Malik, A.; Rehman, F.U.; Shah, K.U.; Naz, S.S.; Qaisar, S. Hemostatic strategies for uncontrolled bleeding: A comprehensive review. J. Biomed. Mater. Res. 2021, 109, 1465–1477. [Google Scholar] [CrossRef] [PubMed]
- Landmann, H. Studies on the mechanism of action of synthetic antifibrinolytics. Thromb. Haemost. 1973, 29, 253–275. [Google Scholar] [CrossRef]
- Fraczyk, J.; Kaminski, Z.J.; Katarzynska, J.; Kolesinska, B. 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium Toluene-4-sulfonate (DMT/NMM/TsO─) universal coupling reagent for synthesis in solution. Helv. Chim. Acta 2018, 101, e1700187. [Google Scholar] [CrossRef] [Green Version]
- Kolesinska, B.; Rozniakowski, K.K.; Fraczyk, J.; Relich, I.; Papini, A.M.; Kaminski, Z.J. The effect of counterion and tertiary amine on th eefficiency of N-triazinylammonium sulfonates in solution and solid-phase peptide synthesis. Eur. J. Org. Chem. 2015, 2015, 401–408. [Google Scholar] [CrossRef]
- Frappé, P.; Cogneau, J.; Gaboreau, Y.; Abenhaim, N.; Bayen, M.; Guichard, C.; Jacquet, J.-P.; Lacoin, F.; Liebart, S.; Bertoletti, L.; et al. Anticoagulants’ safety and effectiveness in general practice: A nationwide prospective cohort study. Ann. Fam. Med. 2020, 18, 131–138. [Google Scholar] [CrossRef]
- Roberti, R.; Iannone, L.F.; Palleria, C.; Curcio, A.; Rossi, M.; Sciacqua, A.; Armentaro, G.; Vero, A.; Manti, A.; Cassano, V.; et al. Direct oral anticoagulants: From randomized clinical trials to real-world clinical practice. Front. Pharmacol. 2021, 12, 684638. [Google Scholar] [CrossRef]
- Royston, D. The current place of aprotinin in the management of bleeding. Anaesthesia 2015, 70, 46–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuda, Y.; Hidaka, K.; Hojo, K.; Okada, Y. Exploration of active site-directed plasmin inhibitors: Beyond tranexamic acid. Processes 2021, 9, 329. [Google Scholar] [CrossRef]
- Al-Horani, R.A.; Desai, R.A. Recent advances on plasmin inhibitors for the treatment of fibrinolysis-related disorders. Med. Res. Rev. 2014, 34, 1168–1216. [Google Scholar] [CrossRef] [PubMed]
- Afosah, D.K.; Al-Horani, R.A.; Sankaranarayanan, N.V.; Desai, U.R. Potent, selective, allosteric inhibition of human plasmin by sulfated non-saccharide glycosaminoglycan mimetics. J. Med. Chem. 2017, 60, 641–657. [Google Scholar] [CrossRef]
- Kolodziejczyk–Czepas, J.; Pasiński, B.; Ponczek, M.B.; Moniuszko-Szajwaj, B.; Kowalczyk, M.; Pecio, Ł.; Nowak, P.; Stochmal, A. Bufadienolides from Kalanchoe daigremontiana modulate the enzymatic activity of plasmin—In vitro and in silico analyses. Int. J. Biol. Macromol. 2018, 120, 1591–1600. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, W.; Yamamoto, H. Tantalum-catalyzed amidation of amino acid homologues. J. Am. Chem. Soc. 2019, 141, 18926–18931. [Google Scholar] [CrossRef] [PubMed]
- Vong, K.K.H.; Maeda, S.; Tanaka, K. Propargyl-assisted selective amidation applied in C-terminal gycine peptide conjugation. Chem. Eur. J. 2016, 22, 18865–18872. [Google Scholar] [CrossRef] [PubMed]
- Midura-Nowaczek, K.; Bruzgo, I.; Popławski, J.; Roszkowska–Jakimiec, W.; Worowski, K. Synthesis and activity of N alpha-(epsilon-aminocaproyl)-alanines and N epsilon-(epsilon-aminocaproyl)-caproicacid. Acta Pol. Pharm. 1995, 52, 509–513. [Google Scholar]
- Frączyk, J.; Walczak, M.; Szymański, Ł.; Kolaciński, Z.; Wrzosek, H.; Majsterek, I.; Przybylewska-Sygut, K.; Kamiński, Z.J. Carbon nanotubes functionalized with folic acid attached via biomimetic peptide linker. Nanomedicine 2017, 12, 2161–2182. [Google Scholar] [CrossRef] [PubMed]
- Kolodziejczyk-Czepas, J.; Sieradzka, M.; Moniuszko-Szajwaj, B.; Nowak, P.; Oleszek, W.; Stochmal, A. Phenolic fractions from nine Trifolium species modulate the coagulant properties of blood plasma in vitro without cytotoxicity towards blood cells. J. Pharm. Pharmacol. 2018, 70, 413–425. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Sample | Concentration [mg/L] | VmaxC (of Fibrin Polymerization) | Amax | VmaxF (of Fibrin Clot Lysis) |
|---|---|---|---|---|
| Control Plasma | 0 | 100.00 % | 100.00% | 100.00% |
| 1 | 10 | 106.12 ± 4.28 ** | 105.98 ± 4.31 * | 99.08 ± 9.93 |
| 25 | 105.66 ± 6.27 | 107.24 ± 8.91 | 87.32 ± 20.27 | |
| 50 | 104.20 ± 3.85 * | 104.62 ± 9.46 | 95.58 ± 15.97 | |
| 2 | 10 | 103.26 ± 3.98 | 105.29 ± 6.41 | 75.81 ± 17.04 * |
| 25 | 106.30 ± 3.60 ** | 110.74 ± 4.86 * | 79.60 ± 10.55 * | |
| 50 | 105.59 ± 3.74 ** | 110.77 ± 9.85 * | 85.69 ± 8.80 * | |
| 3 | 10 | 105.61 ± 7.06 | 107.21 ± 6.90 * | 93.23 ± 16.83 |
| 25 | 103.88 ± 9.73 | 104.49 ± 9.80 | 97.05 ± 24.39 | |
| 50 | 109.78 ± 6.46 * | 112.98 ± 8.40 * | 102.82 ± 20.01 | |
| 4 | 10 | 107.93 ± 7.23 * | 115.37 ± 8.73 * | 81.36 ± 21.07 * |
| 25 | 105.85 ± 7.85 | 114.07 ± 15.71 | 70.35 ± 16.30 * | |
| 50 | 105.89 ± 5.64 | 109.29 ± 9.02 * | 55.95 ± 10.51 * | |
| 5 | 10 | 105.80 ± 2.97 ** | 106.44 ± 1.78 * | 90.08 ± 13.68 |
| 25 | 105.88 ± 3.76 ** | 107.12 ± 4.65 * | 85.12 ± 16.98 * | |
| 50 | 109.83 ± 1.46 *** | 113.43 ± 7.86 * | 89.46 ± 10.48 * | |
| 6 | 10 | 104.68 ± 3.05 * | 105.45 ± 5.46 | 98.41 ± 14.71 |
| 25 | 103.10 ± 6.82 | 102.41 ± 5.56 | 100.06 ± 21.96 | |
| 50 | 101.11 ± 4.77 | 102.15 ± 2.46 | 91.08 ± 15.49 | |
| 7 | 10 | 106.86 ± 9.49 | 115.64 ± 11.61 * | 66.18 ± 21.32 * |
| 25 | 104.39 ± 8.60 | 120.06 ± 25.63 * | 62.99 ± 16.99 * | |
| 50 | 111.45 ± 8.07 ** | 113.54 ± 13.76 * | 87.27 ± 8.04 | |
| 8 | 10 | 101.96 ± 6.62 | 104.83 ± 2.83 * | 82.96 ± 12.49 * |
| 25 | 100.98 ± 6.60 | 100.18 ± 3.77 | 96.07 ± 14.56 | |
| 50 | 105.67 ± 8.34 | 108.64 ± 11.94 | 66.90 ± 23.22 * | |
| TA | 10 | 101.38 ± 7.89 | 106.67 ± 3.79 * | total inhibition |
| 25 | 104.75 ± 9.98 | 105.87 ± 6.23 | total inhibition | |
| 50 | 104.85 ± 12.06 | 106.64 ± 4.65 * | total inhibition |
| Amide | Hemolysis Assay | Blood Plasma Clotting Assays | |||||
|---|---|---|---|---|---|---|---|
| c [mg/L] | Time [min] | Hemolysis [%] * | c [mg/L] | PT Time [% of Control] | aPTT Time [% of Control] | TT Time [% of Control] | |
| 1 | 5 | 45 | 0.0 | 10 | 96.3 ± 2.5 | 108.3 ± 11.9 | 95.9 ± 6.3 |
| 120 | 0.56 | ||||||
| 240 | 0.42 | ||||||
| 50 | 45 | 0.5 | 25 | 95.0 ± 2.9 | 105.1 ± 7.7 | 96.1 ± 6.8 | |
| 120 | 0.18 | ||||||
| 240 | 0.35 | ||||||
| 500 | 45 | 0.27 | 50 | 95.7 ± 2.2 | 101.9 ± 9.3 | 94.3 ± 8.3 | |
| 120 | 0.59 | ||||||
| 240 | 0.58 | ||||||
| 2 | 5 | 45 | 0.41 | 10 | 97.0 ± 4.8 | 98.5 ± 8.0 | 99.0 ± 1.8 |
| 120 | 0.21 | ||||||
| 240 | 0.65 | ||||||
| 50 | 45 | 0.11 | 25 | 96.1 ± 2.2 | 101.0 ± 13.1 | 98.9 ± 4.7 | |
| 120 | 0.74 | ||||||
| 240 | 0.30 | ||||||
| 500 | 45 | 1.03 | 50 | 95.5 ± 2.5 | 101.9 ± 14.0 | 95.9 ± 6.8 | |
| 120 | 0.21 | ||||||
| 240 | 0.43 | ||||||
| 3 | 5 | 45 | 0.44 | 10 | 94.9 ± 2.2 | 94.8 ± 4.5 | 100.1 ± 2.6 |
| 120 | 0.74 | ||||||
| 240 | 0.41 | ||||||
| 50 | 45 | 0.0 | 25 | 95.2 ± 3.0 | 94.7 ± 4.1 | 100.8 ± 6.9 | |
| 120 | 0.0 | ||||||
| 240 | 0.26 | ||||||
| 500 | 45 | 0.47 | 50 | 97.0 ± 3.6 | 96.3 ± 2.8 | 97.2 ± 4.3 | |
| 120 | 0.65 | ||||||
| 240 | 0.23 | ||||||
| 4 | 5 | 45 | 1.0 | 10 | 97.3 ± 4.2 | 98.1 ± 5.2 | 96.3 ± 6.9 |
| 120 | 0.74 | ||||||
| 240 | 0.27 | ||||||
| 50 | 45 | 0.0 | 25 | 95.4 ± 2.3 | 97.4 ± 5.4 | 95.5 ± 8.8 | |
| 120 | 0.71 | ||||||
| 240 | 0.32 | ||||||
| 500 | 45 | 0.38 | 50 | 93.7 ± 3.0 | 99.9 ± 6.0 | 97.6 ± 5.0 | |
| 120 | 0.21 | ||||||
| 240 | 0.22 | ||||||
| 5 | 5 | 45 | 0.0 | 10 | 95.0 ± 10.5 | 98.8 ± 5.8 | 99.1 ± 6.1 |
| 120 | 0.56 | ||||||
| 240 | 0.42 | ||||||
| 50 | 45 | 0.5 | 25 | 92.0 ± 8.5 | 101.1 ± 4.9 | 99.0 ± 4.7 | |
| 120 | 0.18 | ||||||
| 240 | 0.35 | ||||||
| 500 | 45 | 0.27 | 50 | 93.0 ± 8.3 | 100.8 ± 2.6 | 97.8 ± 3.3 | |
| 120 | 0.59 | ||||||
| 240 | 0.58 | ||||||
| 6 | 5 | 45 | 0.38 | 10 | 96.1 ± 4.4 | 96.1 ± 4.2 | 92.5 ± 3.0 |
| 120 | 0.56 | ||||||
| 240 | 0.19 | ||||||
| 50 | 45 | 0.0 | 25 | 95.8 ± 4.3 | 97.0 ± 3.5 | 92.3 ± 2.5 | |
| 120 | 0.11 | ||||||
| 240 | 0.2 | ||||||
| 500 | 45 | 0.41 | 50 | 93.5 ± 7.1 | 98.3 ± 3.6 | 92.8 ± 4.2 | |
| 120 | 0.68 | ||||||
| 240 | 0.31 | ||||||
| 7 | 5 | 45 | 0.0 | 10 | 95.4 ± 7.4 | 98.1 ± 4.6 | 97.2 ± 3.3 |
| 120 | 0.21 | ||||||
| 240 | 0.21 | ||||||
| 50 | 45 | 0.41 | 25 | 94.3 ± 9.7 | 99.3 ± 3.3 | 97.5 ± 2.5 | |
| 120 | 0.97 | ||||||
| 240 | 0.64 | ||||||
| 50 | 45 | 0.0 | 50 | 95.3 ± 8.3 | 100.6 ± 4.9 | 96.9 ± 1.7 | |
| 120 | 0.23 | ||||||
| 240 | 0.38 | ||||||
| 8 | 5 | 45 | 0.08 | 10 | 93.0 ± 2.2 | 95.1 ± 7.5 | 100.0 ± 7.0 |
| 120 | 0.74 | ||||||
| 240 | 0.36 | ||||||
| 50 | 45 | 0.44 | 25 | 95.5 ± 4.3 | 97.7 ± 5.9 | 94.3 ± 1.3 | |
| 120 | 0.71 | ||||||
| 240 | 0.3 | ||||||
| 50 | 45 | 0.0 | 50 | 97.2 ± 3.2 | 95.2 ± 7.3 | 92.4 ± 5.2 | |
| 120 | 0.47 | ||||||
| 240 | 0.37 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banach, L.; Janczewski, L.; Kajdanek, J.; Milowska, K.; Kolodziejczyk-Czepas, J.; Galita, G.; Rozpedek-Kaminska, W.; Kucharska, E.; Majsterek, I.; Kolesinska, B. Synthesis and Hemostatic Activity of New Amide Derivatives. Molecules 2022, 27, 2271. https://doi.org/10.3390/molecules27072271
Banach L, Janczewski L, Kajdanek J, Milowska K, Kolodziejczyk-Czepas J, Galita G, Rozpedek-Kaminska W, Kucharska E, Majsterek I, Kolesinska B. Synthesis and Hemostatic Activity of New Amide Derivatives. Molecules. 2022; 27(7):2271. https://doi.org/10.3390/molecules27072271
Chicago/Turabian StyleBanach, Lukasz, Lukasz Janczewski, Jakub Kajdanek, Katarzyna Milowska, Joanna Kolodziejczyk-Czepas, Grzegorz Galita, Wioletta Rozpedek-Kaminska, Ewa Kucharska, Ireneusz Majsterek, and Beata Kolesinska. 2022. "Synthesis and Hemostatic Activity of New Amide Derivatives" Molecules 27, no. 7: 2271. https://doi.org/10.3390/molecules27072271
APA StyleBanach, L., Janczewski, L., Kajdanek, J., Milowska, K., Kolodziejczyk-Czepas, J., Galita, G., Rozpedek-Kaminska, W., Kucharska, E., Majsterek, I., & Kolesinska, B. (2022). Synthesis and Hemostatic Activity of New Amide Derivatives. Molecules, 27(7), 2271. https://doi.org/10.3390/molecules27072271