
Computational Analysis and Biological Activities of Oxyresveratrol Analogues, the Putative Cyclooxygenase-2 Inhibitors

 ,
, 
Abstract
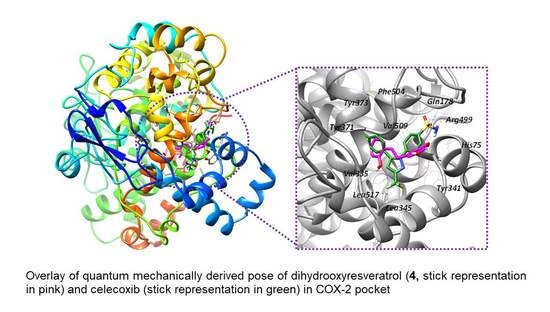
:
1. Introduction
2. Results and Discussion
2.1. Binding Affinity of COX-2/Celecoxib Complex
2.2. Validation of Docking Protocols
2.3. Molecular Docking of Oxyresveratrol Analogues
2.4. Biological Activity of Oxyresveratrol Analogues
2.5. Rescoring Oxyresveratrol Analogues by Quantum Mechanical (QM) Binding Energy
2.6. Fluorinated Analogues
2.7. Atomic Charges of Celecoxib and Oxyresveratrol Analogues
3. Statistical Analysis
4. Conclusions
5. Experimental Section
5.1. Chemistry
5.1.1. Isolation of Oxyresveratrol from A. lacucha
5.1.2. General Procedure for the Synthesis of Compound 2
5.1.3. General Procedure for the Synthesis of Compounds 4 and 5
5.1.4. General Procedure for the Synthesis of Compounds 3 and 6
5.2. Characterization
5.3. Measurement of Biological Activities
5.3.1. COX-2 Inhibitory Activity
5.3.2. Cytotoxicity Assay
6. Computational Methods
6.1. Preparation of Enzyme Receptor
6.2. Computational Models of Ligand Structures
6.3. Molecular Docking Protocols
6.4. Computation of QM Energy and Binding Energy (BE)
6.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Ricciotti, E.; FitzGerald, G.A. Prostaglandins and Inflammation. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 986–1000. [Google Scholar] [CrossRef] [PubMed]
- Rouzer, C.A.; Marnett, L.J. Non-redundant functions of cyclooxygenases: Oxygenation of endocannabinoids. J. Biol. Chem. 2008, 283, 8065–8069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulmacz, R.J.; Wang, L.-H. Comparison of Hydroperoxide Initiator Requirements for the Cyclooxygenase Activities of Prostaglandin H Synthase-1 and -2. J. Biol. Chem. 1995, 270, 24019–24023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warner, T.D.; Mitchell, J.A. Cyclooxygenases: New forms, new inhibitors, and lessons from the clinic. FASEB J. 2004, 18, 790–804. [Google Scholar] [CrossRef]
- Knights, K.M.; Mangoni, A.A.; Miners, J.O. Defining the COX inhibitor selectivity of NSAIDs: Implications for understanding toxicity. Expert Rev. Clin. Pharmacol. 2010, 3, 769–776. [Google Scholar] [CrossRef]
- Williams, C.S.; Mann, M.; DuBois, R.N. The role of cyclooxygenases in inflammation, cancer, and development. Oncogene 1999, 18, 7908–7916. [Google Scholar] [CrossRef] [Green Version]
- Dubois, R.N.; Abramson, S.B.; Crofford, L.; Gupta, R.A.; Simon, L.S.; Van De Putte, L.B.; Lipsky, P.E. Cyclooxygenase in biology and disease. FASEB J. 1998, 12, 1063–1073. [Google Scholar] [CrossRef] [Green Version]
- Dvorakova, M.; Landa, P. Anti-inflammatory activity of natural stilbenoids: A review. Pharmacol. Res. 2017, 124, 126–145. [Google Scholar] [CrossRef]
- Machado, N.D.; Fernández, M.A.; Díaz, D.D. Recent Strategies in Resveratrol Delivery Systems. ChemPlusChem 2019, 84, 951–973. [Google Scholar] [CrossRef]
- Limongelli, V.; Bonomi, M.; Marinelli, L.; Gervasio, F.L.; Cavalli, A.; Novellino, E.; Parrinello, M. Molecular basis of cyclooxygenase enzymes (COXs) selective inhibition. Proc. Natl. Acad. Sci. USA 2010, 107, 5411. [Google Scholar] [CrossRef] [Green Version]
- Anita-Marie, R.; Nathalie, L.; Clotilde, F.; Jean-Francois, Z.; Matthieu, M.; Maite Sylla-Iyarreta, V. Update on COX-2 Selective Inhibitors: Chemical Classification, Side Effects and their Use in Cancers and Neuronal Diseases. Curr. Top. Med. Chem. 2017, 17, 2935–2956. [Google Scholar]
- White, W.B.; Faich, G.; Borer, J.S.; Makuch, R.W. Cardiovascular thrombotic events in arthritis trials of the cyclooxygenase-2 inhibitor celecoxib**The investigators had full access to the data and complete control over the design, conduct analysis, interpretation, and description of the study. The sponsor of the study had no role in study design, data collection, data analysis, data interpretation, or in writing the report. The views expressed are those of the investigators alone. Am. J. Cardiol. 2003, 92, 411–418. [Google Scholar] [PubMed]
- Mahesh, G.; Anil Kumar, K.; Reddanna, P. Overview on the Discovery and Development of Anti-Inflammatory Drugs: Should the Focus Be on Synthesis or Degradation of PGE(2)? J. Inflamm. Res. 2021, 14, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Viliam, B.; Karel, S.; Jan, H.; Veronika, T. Anti-inflammatory Natural Prenylated Phenolic Compounds—Potential Lead Substances. Curr. Med. Chem. 2018, 25, 1094–1159. [Google Scholar]
- Liu, J.-Q.; Lian, C.-L.; Hu, T.-Y.; Wang, C.-F.; Xu, Y.; Xiao, L.; Liu, Z.-Q.; Qiu, S.-Q.; Cheng, B.-H. Two new farnesyl phenolic compounds with anti-inflammatory activities from Ganoderma duripora. Food Chem. 2018, 263, 155–162. [Google Scholar] [CrossRef]
- Lescano, C.H.; Freitas de Lima, F.; Mendes-Silvério, C.B.; Justo, A.F.O.; da Silva Baldivia, D.; Vieira, C.P.; Sanjinez-Argandoña, E.J.; Cardoso, C.A.L.; Mónica, F.Z.; Pires de Oliveira, I. Effect of Polyphenols from Campomanesia adamantium on Platelet Aggregation and Inhibition of Cyclooxygenases: Molecular Docking and in Vitro Analysis. Front. Pharmacol. 2018, 9, 617. [Google Scholar] [CrossRef] [Green Version]
- Ayertey, F.; Ofori-Attah, E.; Antwi, S.; Amoa-Bosompem, M.; Djameh, G.; Lartey, N.L.; Ohashi, M.; Kusi, K.A.; Appiah, A.A.; Appiah-Opong, R.; et al. Anti-inflammatory activity and mechanism of action of ethanolic leaf extract of Morinda lucida Benth. J. Tradit. Complementary Med. 2021, 11, 249–258. [Google Scholar] [CrossRef]
- Abubakar, S.; Al-Mansoub, M.A.; Murugaiyah, V.; Chan, K.-L. The phytochemical and anti-inflammatory studies of Dillenia suffruticosa leaves. Phytother. Res. 2019, 33, 660–675. [Google Scholar] [CrossRef]
- Adebayo, S.A.; Shai, L.J.; Eloff, J.N. First isolation of glutinol and a bioactive fraction with good anti-inflammatory activity from n-hexane fraction of Peltophorum africanum leaf. Asian Pac. J. Trop. Med. 2017, 10, 42–46. [Google Scholar] [CrossRef]
- Ghanta, P.; Doble, M.; Ramaiah, B. Alkaloids of Adhatoda vasica Nees. as potential inhibitors of cyclooxygenases—An in-silico study. J. Biomol. Struct. Dyn. 2021, 1–11. [Google Scholar] [CrossRef]
- Yang, L.; Zhu, Y.; He, Z.; Zhang, T.; Xiao, Z.; Xu, R.; He, J. Plantanone D, a new rare methyl-flavonoid from the flowers of Hosta plantaginea with anti-inflammatory and antioxidant activities. Nat. Prod. Res. 2021, 35, 4331–4337. [Google Scholar] [CrossRef] [PubMed]
- Hanáková, Z.; Hošek, J.; Kutil, Z.; Temml, V.; Landa, P.; Vaněk, T.; Schuster, D.; Dall’Acqua, S.; Cvačka, J.; Polanský, O.; et al. Anti-inflammatory Activity of Natural Geranylated Flavonoids: Cyclooxygenase and Lipoxygenase Inhibitory Properties and Proteomic Analysis. J. Nat. Prod. 2017, 80, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Brglez Mojzer, E.; Knez Hrnčič, M.; Škerget, M.; Knez, Ž.; Bren, U. Polyphenols: Extraction Methods, Antioxidative Action, Bioavailability and Anticarcinogenic Effects. Molecules 2016, 21, 901. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Cai, L.; Udeani, G.O.; Slowing, K.V.; Thomas, C.F.; Beecher, C.W.W.; Fong, H.H.S.; Farnsworth, N.R.; Kinghorn, A.D.; Mehta, R.G.; et al. Cancer Chemopreventive Activity of Resveratrol, a Natural Product Derived from Grapes. Science 1997, 275, 218–220. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Kim, S.; Kwon, O.-K.; Oh, S.-R.; Lee, H.-K.; Ahn, K. Anti-inflammatory and anti-asthmatic effects of resveratrol, a polyphenolic stilbene, in a mouse model of allergic asthma. Int. Immunopharmacol. 2009, 9, 418–424. [Google Scholar] [CrossRef]
- Bernard, P.; Berthon, J.Y. Resveratrol: An original mechanism on tyrosinase inhibition. Int. J. Cosmet. Sci. 2000, 22, 219–226. [Google Scholar] [CrossRef]
- Fang, S.-C.; Hsu, C.-L.; Yen, G.-C. Anti-inflammatory Effects of Phenolic Compounds Isolated from the Fruits of Artocarpus heterophyllus. J. Agric. Food Chem. 2008, 56, 4463–4468. [Google Scholar] [CrossRef]
- Powell, R.G.; Bajaj, R.; McLaughlin, J.L. Bioactive Stilbenes of Scirpus maritimus. J. Nat. Prod. 1987, 50, 293–296. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Spina, M.G.; Lorenz, P.; Ebmeyer, U.; Wolf, G.; Horn, T.F.W. Oxyresveratrol (trans-2,3′,4,5′-tetrahydroxystilbene) is neuroprotective and inhibits the apoptotic cell death in transient cerebral ischemia. Brain Res. 2004, 1017, 98–107. [Google Scholar] [CrossRef]
- Chatsumpun, N.; Chuanasa, T.; Sritularak, B.; Lipipun, V.; Jongbunprasert, V.; Ruchirawat, S.; Ploypradith, P.; Likhitwitayawuid, K. Oxyresveratrol: Structural Modification and Evaluation of Biological Activities. Molecules 2016, 21, 489. [Google Scholar] [CrossRef] [Green Version]
- Likhitwitayawuid, K.; Sornsute, A.; Sritularak, B.; Ploypradith, P. Chemical transformations of oxyresveratrol (trans-2,4,3′,5′-tetrahydroxystilbene) into a potent tyrosinase inhibitor and a strong cytotoxic agent. Bioorganic Med. Chem. Lett. 2006, 16, 5650–5653. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-Q.; Li, Z.-L.; Zhao, W.-J.; Wen, R.-X.; Meng, Q.-W.; Zeng, Y. Synthesis of stilbene derivatives with inhibition of SARS coronavirus replication. Eur. J. Med. Chem. 2006, 41, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Likhitwitayawuid, K. Oxyresveratrol: Sources, Productions, Biological Activities, Pharmacokinetics, and Delivery Systems. Molecules 2021, 26, 4212. [Google Scholar] [CrossRef] [PubMed]
- Kutil, Z.; Kvasnicova, M.; Temml, V.; Schuster, D.; Marsik, P.; Cusimamani, E.F.; Lou, J.-D.; Vanek, T.; Landa, P. Effect of Dietary Stilbenes on 5-Lipoxygenase and Cyclooxygenases Activities In Vitro. Int. J. Food Prop. 2015, 18, 1471–1477. [Google Scholar] [CrossRef]
- Chung, K.-O.; Kim, B.-Y.; Lee, M.-H.; Kim, Y.-R.; Chung, H.-Y.; Park, J.-H.; Moon, J.-O. In-vitro and in-vivo anti-inflammatory effect of oxyresveratrol from Morus alba L. J. Pharm. Pharmacol. 2010, 55, 1695–1700. [Google Scholar] [CrossRef]
- Aziz, R.S.; Siddiqua, A.; Shahzad, M.; Shabbir, A.; Naseem, N. Oxyresveratrol ameliorates ethanol-induced gastric ulcer via downregulation of IL-6, TNF-α, NF-ĸB, and COX-2 levels, and upregulation of TFF-2 levels. Biomed. Pharmacother. 2019, 110, 554–560. [Google Scholar] [CrossRef]
- Sharma, V.; Bhatia, P.; Alam, O.; Javed Naim, M.; Nawaz, F.; Ahmad Sheikh, A.; Jha, M. Recent advancement in the discovery and development of COX-2 inhibitors: Insight into biological activities and SAR studies (2008–2019). Bioorganic Chem. 2019, 89, 103007. [Google Scholar] [CrossRef]
- McCormack, P.L. Celecoxib. Drugs 2011, 71, 2457–2489. [Google Scholar] [CrossRef]
- Ferreira, L.G.; Dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular Docking and Structure-Based Drug Design Strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for molecular docking: A review. Biophys. Rev. 2017, 9, 91–102. [Google Scholar] [CrossRef]
- Guedes, I.A.; de Magalhães, C.S.; Dardenne, L.E. Receptor–ligand molecular docking. Biophys. Rev. 2014, 6, 75–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuriev, E.; Holien, J.; Ramsland, P.A. Improvements, trends, and new ideas in molecular docking: 2012–2013 in review. J. Mol. Recognit. 2015, 28, 581–604. [Google Scholar] [CrossRef] [PubMed]
- Adeniyi, A.A.; Soliman, M.E.S. Implementing QM in docking calculations: Is it a waste of computational time? Drug Discov. Today 2017, 22, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-C. Beware of docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef]
- Zhou, T.; Huang, D.; Caflisch, A. Quantum Mechanical Methods for Drug Design. Curr. Top. Med. Chem. 2010, 10, 33–45. [Google Scholar] [CrossRef] [Green Version]
- Mazanetz, M.P. Quantum mechanical applications in drug discovery. In In Silico Drug Discovery and Design; Future Medicine: London, UK, 2013; pp. 64–79. [Google Scholar]
- Raha, K.; Peters, M.B.; Wang, B.; Yu, N.; Wollacott, A.M.; Westerhoff, L.M.; Merz, K.M. The role of quantum mechanics in structure-based drug design. Drug Discov. Today 2007, 12, 725–731. [Google Scholar] [CrossRef]
- Atlam, F.M.; Awad, M.K.; El-Bastawissy, E.A. Computational simulation of the effect of quantum chemical parameters on the molecular docking of HMG-CoA reductase drugs. J. Mol. Struct. 2014, 1075, 311–326. [Google Scholar] [CrossRef]
- Picot, D.; Loll, P.J.; Garavito, R.M. The X-ray crystal structure of the membrane protein prostaglandin H2 synthase-1. Nature 1994, 367, 243–249. [Google Scholar] [CrossRef]
- Llorens, O.; Perez, J.J.; Palomer, A.; Mauleon, D. Differential binding mode of diverse cyclooxygenase inhibitors. J. Mol. Graph. Model. 2002, 20, 359–371. [Google Scholar] [CrossRef]
- Wang, J.L.; Carter, J.; Kiefer, J.R.; Kurumbail, R.G.; Pawlitz, J.L.; Brown, D.; Hartmann, S.J.; Graneto, M.J.; Seibert, K.; Talley, J.J. The novel benzopyran class of selective cyclooxygenase-2 inhibitors-part I: The first clinical candidate. Bioorganic Med. Chem. Lett. 2010, 20, 7155–7158. [Google Scholar] [CrossRef]
- Ghareb, N.; Elshihawy, H.A.; Abdel-Daim, M.M.; Helal, M.A. Novel pyrazoles and pyrazolo[1,2-a]pyridazines as selective COX-2 inhibitors; Ultrasound-assisted synthesis, biological evaluation, and DFT calculations. Bioorganic Med. Chem. Lett. 2017, 27, 2377–2383. [Google Scholar] [CrossRef] [PubMed]
- Michaux, C.; de Leval, X.; Julémont, F.; Dogné, J.-M.; Pirotte, B.; Durant, F. Structure-based pharmacophore of COX-2 selective inhibitors and identification of original lead compounds from 3D database searching method. Eur. J. Med. Chem. 2006, 41, 1446–1455. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, N.; Aparoy, P. Deciphering the mechanism behind the varied binding activities of COXIBs through Molecular Dynamic Simulations, MM-PBSA binding energy calculations and per-residue energy decomposition studies. J. Biomol. Struct. Dyn. 2017, 35, 868–882. [Google Scholar] [CrossRef] [PubMed]
- Coy-Barrera, E. Discrimination of Naturally-Occurring 2-Arylbenzofurans as Cyclooxygenase-2 Inhibitors: Insights into the Binding Mode and Enzymatic Inhibitory Activity. Biomolecules 2020, 10, 176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [Green Version]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein–ligand docking using GOLD. Proteins Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking11Edited by F. E. Cohen. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [Green Version]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical Scoring Functions for Advanced Protein−Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Sapundzhi, F.; Prodanova, K.; Lazarova, M. Survey of the scoring functions for protein-ligand docking. AIP Conf. Proc. 2019, 2172, 100008. [Google Scholar]
- Shukla, S.; Bafna, K.; Sundar, D.; Thorat, S.S. The Bitter Barricading of Prostaglandin Biosynthesis Pathway: Understanding the Molecular Mechanism of Selective Cyclooxygenase-2 Inhibition by Amarogentin, a Secoiridoid Glycoside from Swertia chirayita. PLoS ONE 2014, 9, e90637. [Google Scholar] [CrossRef] [Green Version]
- Shrivastava, N.; Joshi, J.; Sehgal, N.; Kumar, I.P. Cyclooxygenase-2 identified as a potential target for novel radiomodulator scopolamine methyl bromide: An in silico study. Inform. Med. Unlocked 2017, 9, 18–25. [Google Scholar] [CrossRef]
- Maicheen, C.; Phosrithong, N.; Ungwitayatorn, J. Biological activity evaluation and molecular docking study of chromone derivatives as cyclooxygenase-2 inhibitors. Med. Chem. Res. 2017, 26, 662–671. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Xiao, H.; Li, T.; Sun, X.-L.; Wan, W.-M.; Bao, H.; Qian, Q.; Chen, Q. Unpredicted Concentration-Dependent Sensory Properties of Pyrene-Containing NBN-Doped Polycyclic Aromatic Hydrocarbons. Molecules 2022, 27, 327. [Google Scholar] [CrossRef]
- Ueno, H.; Maruyama, A.; Miyake, M.; Nakao, E.; Nakao, K.; Umezu, K.; Nitta, I. Synthesis and evaluation of antiinflammatory activities of a series of corticosteroid 17.alpha.-esters containing a functional group. J. Med. Chem. 1991, 34, 2468–2473. [Google Scholar] [CrossRef]
- Murias, M.; Handler, N.; Erker, T.; Pleban, K.; Ecker, G.; Saiko, P.; Szekeres, T.; Jäger, W. Resveratrol analogues as selective cyclooxygenase-2 inhibitors: Synthesis and structure–activity relationship. Bioorganic Med. Chem. 2004, 12, 5571–5578. [Google Scholar] [CrossRef]
- Alloatti, D.; Giannini, G.; Cabri, W.; Lustrati, I.; Marzi, M.; Ciacci, A.; Gallo, G.; Tinti, M.O.; Marcellini, M.; Riccioni, T.; et al. Synthesis and Biological Activity of Fluorinated Combretastatin Analogues. J. Med. Chem. 2008, 51, 2708–2721. [Google Scholar] [CrossRef]
- Rivera, H.; Morales-Ríos, M.S.; Bautista, W.; Shibayama, M.; Tsutsumi, V.; Muriel, P.; Pérez-Álvarez, V. A novel fluorinated stilbene exerts hepatoprotective properties in CCl4-induced acute liver damage. Can. J. Physiol. Pharmacol. 2011, 89, 759–766. [Google Scholar] [CrossRef]
- Shah, P.; Westwell, A.D. The role of fluorine in medicinal chemistry. J. Enzym. Inhib. Med. Chem. 2007, 22, 527–540. [Google Scholar] [CrossRef] [Green Version]
- Kores, K.; Lešnik, S.; Bren, U.; Janežič, D.; Konc, J. Discovery of Novel Potential Human Targets of Resveratrol by Inverse Molecular Docking. J. Chem. Inf. Model. 2019, 59, 2467–2478. [Google Scholar] [CrossRef]
- King, E.; Aitchison, E.; Li, H.; Luo, R. Recent Developments in Free Energy Calculations for Drug Discovery. Front. Mol. Biosci. 2021, 8, 712085. [Google Scholar] [CrossRef] [PubMed]
- Furlan, V.; Bren, U. Insight into Inhibitory Mechanism of PDE4D by Dietary Polyphenols Using Molecular Dynamics Simulations and Free Energy Calculations. Biomolecules 2021, 11, 479. [Google Scholar] [CrossRef] [PubMed]
- Duangdee, N.; Chamboonchu, N.; Kongkiatpaiboon, S.; Prateeptongkum, S. Quantitative 1HNMR spectroscopy for the determination of oxyresveratrol in Artocarpus lacucha heartwood. Phytochem. Anal. 2019, 30, 617–622. [Google Scholar] [CrossRef] [PubMed]
- Mongolsuk, S.; Robertson, A.; Towers, R. 429. 2: 4: 3′: 5′-Tetrahydroxystilbene from Artocarpus lakoocha. J. Chem. Soc. 1957, 2231–2233. [Google Scholar] [CrossRef]
- Yan, Y.-M.; Zhang, H.-X.; Liu, H.; Wang, Y.; Wu, J.-B.; Li, Y.-P.; Cheng, Y.-X. (+/−)-Lucidumone, a COX-2 Inhibitory Caged Fungal Meroterpenoid from Ganoderma lucidum. Org. Lett. 2019, 21, 8523–8527. [Google Scholar] [CrossRef]
- Singh, P.; Kaur, S.; Kaur, J.; Singh, G.; Bhatti, R. Rational Design of Small Peptides for Optimal Inhibition of Cyclooxygenase-2: Development of a Highly Effective Anti-Inflammatory Agent. J. Med. Chem. 2016, 59, 3920–3934. [Google Scholar] [CrossRef]
- Mahavorasirikul, W.; Viyanant, V.; Chaijaroenkul, W.; Itharat, A.; Na-Bangchang, K. Cytotoxic activity of Thai medicinal plants against human cholangiocarcinoma, laryngeal and hepatocarcinoma cells in vitro. BMC Complement. Altern. Med. 2010, 10, 55. [Google Scholar] [CrossRef] [Green Version]
- BIOVIA; Discovery Studio Modeling Environment, Release 2017; Dassault Systèmes: San Diego, CA, USA, 2016.
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]
- Jongkon, N.; Gleeson, D.; Gleeson, M.P. Elucidation of the catalytic mechanism of 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase using QM/MM calculations. Org. Biomol. Chem. 2018, 16, 6239–6249. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser interfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Simeon, S.; Jongkon, N.; Chotpatiwetchkul, W.; Gleeson, M.P. Insights into the EGFR SAR of N-phenylquinazolin-4-amine-derivatives using quantum mechanical pairwise-interaction energies. J. Comput.-Aided Mol. Des. 2019, 33, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Jaiyong, P.; Bryce, R.A. Approximate quantum chemical methods for modelling carbohydrate conformation and aromatic interactions: β-cyclodextrin and its adsorption on a single-layer graphene sheet. Phys. Chem. Chem. Phys. 2017, 19, 15346–15355. [Google Scholar] [CrossRef]
- IBM SPSS Statistics for Windows; Version 27.0.; Released 2020; IBM Corp: Armonk, NY, USA, 2020.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | AutoDock | GOLD Fitness Function | QM in Vacuo | QM in Implicit Aqueous Solvation | COX-2 Inhibition | MRC-5 Cytotoxicity | |||
|---|---|---|---|---|---|---|---|---|---|
| Binding Affinity | GoldScore | ChemPLP | BE (Raw) | BE (CP) | BE (Raw) | BE (CP) | IC50/(µM) | CC50/(µM) | |
 | −11.24 | 79.07 | 95.56 | −65.39 | −55.04 | −36.11 | −25.76 | 0.09 ± 0.01 | n.t. * |
 | −7.54 | 55.90 | 68.34 | −77.32 | −68.47 | −33.68 | −24.84 | 14.50 ± 2.04 | 57.48 ± 2.34 |
 | −6.94 | 59.35 | 57.63 | −56.51 | −47.30 | −25.18 | −15.98 | 25.00 ± 2.34 | 118.21 ± 2.69 |
 | −9.21 | 80.20 | 62.08 | −80.67 | −68.72 | −35.36 | −23.42 | 23.00 ± 2.02 | 77.66 ± 3.07 |
 | −6.62 | 56.91 | 74.09 | −85.29 | −75.98 | −47.16 | −37.86 | 11.50 ± 1.54 | 106.02 ± 3.86 |
 | −6.69 | 60.09 | 66.57 | −58.86 | −49.75 | −28.78 | −19.68 | 18.10 ± 2.07 | 130.32 ± 3.04 |
 | −9.26 | 78.62 | 83.45 | −82.35 | −70.34 | −36.89 | −24.88 | 19.00 ± 2.01 | 120.51 ± 2.37 |
 | −6.64 | 58.07 | 67.66 | −87.74 | −78.44 | −47.21 | −37.90 | n.t. * | n.t. * |
 | −6.55 | 63.34 | 62.82 | −55.62 | −44.92 | −23.28 | −12.59 | n.t. * | 53.04 ± 0.22 |
 | −8.82 | 78.15 | 61.22 | −81.38 | −68.65 | −36.78 | −24.04 | n.t. * | 11.69 ± 1.35 |
| Ligand | Celecoxib | Ring B | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| bound | S 0.306 | C4 | −0.539 | −0.060 | 0.111 | 0.642 | −0.308 | 0.154 | 0.082 | 0.021 | 0.170 |
| C1 | −0.059 | 0.090 | −0.280 | 0.425 | −0.796 | −1.064 | −0.014 | −0.630 | −1.538 | ||
| unbound | S 1.514 | C4 | 0.551 | 0.419 | 0.363 | 0.730 | 0.461 | 0.476 | 0.699 | 0.225 | 0.108 |
| C1 | 0.253 | −0.133 | 0.190 | −0.078 | −0.509 | −0.045 | −0.165 | −0.204 | 0.118 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jongkon, N.; Seaho, B.; Tayana, N.; Prateeptongkum, S.; Duangdee, N.; Jaiyong, P. Computational Analysis and Biological Activities of Oxyresveratrol Analogues, the Putative Cyclooxygenase-2 Inhibitors. Molecules 2022, 27, 2346. https://doi.org/10.3390/molecules27072346
Jongkon N, Seaho B, Tayana N, Prateeptongkum S, Duangdee N, Jaiyong P. Computational Analysis and Biological Activities of Oxyresveratrol Analogues, the Putative Cyclooxygenase-2 Inhibitors. Molecules. 2022; 27(7):2346. https://doi.org/10.3390/molecules27072346
Chicago/Turabian StyleJongkon, Nathjanan, Boonwiset Seaho, Ngampuk Tayana, Saisuree Prateeptongkum, Nongnaphat Duangdee, and Panichakorn Jaiyong. 2022. "Computational Analysis and Biological Activities of Oxyresveratrol Analogues, the Putative Cyclooxygenase-2 Inhibitors" Molecules 27, no. 7: 2346. https://doi.org/10.3390/molecules27072346
APA StyleJongkon, N., Seaho, B., Tayana, N., Prateeptongkum, S., Duangdee, N., & Jaiyong, P. (2022). Computational Analysis and Biological Activities of Oxyresveratrol Analogues, the Putative Cyclooxygenase-2 Inhibitors. Molecules, 27(7), 2346. https://doi.org/10.3390/molecules27072346