
Quantification of Structural Integrity and Stability Using Nanograms of Protein by Flow-Induced Dispersion Analysis


Abstract
:1. Introduction
2. Results and Discussion
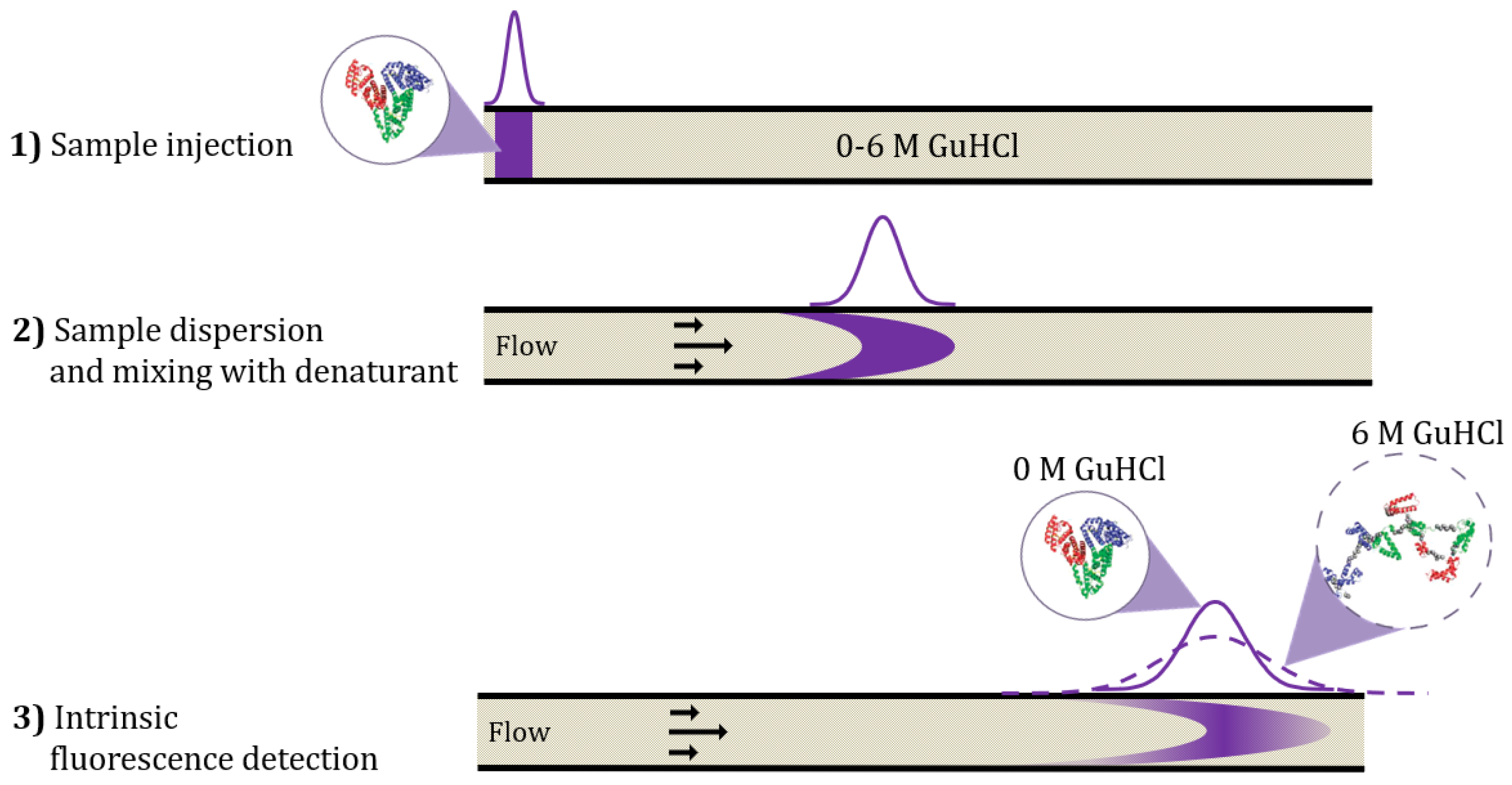
2.1. Assay Development of Capillary Mixing Mode for Protein-Stability Assessment
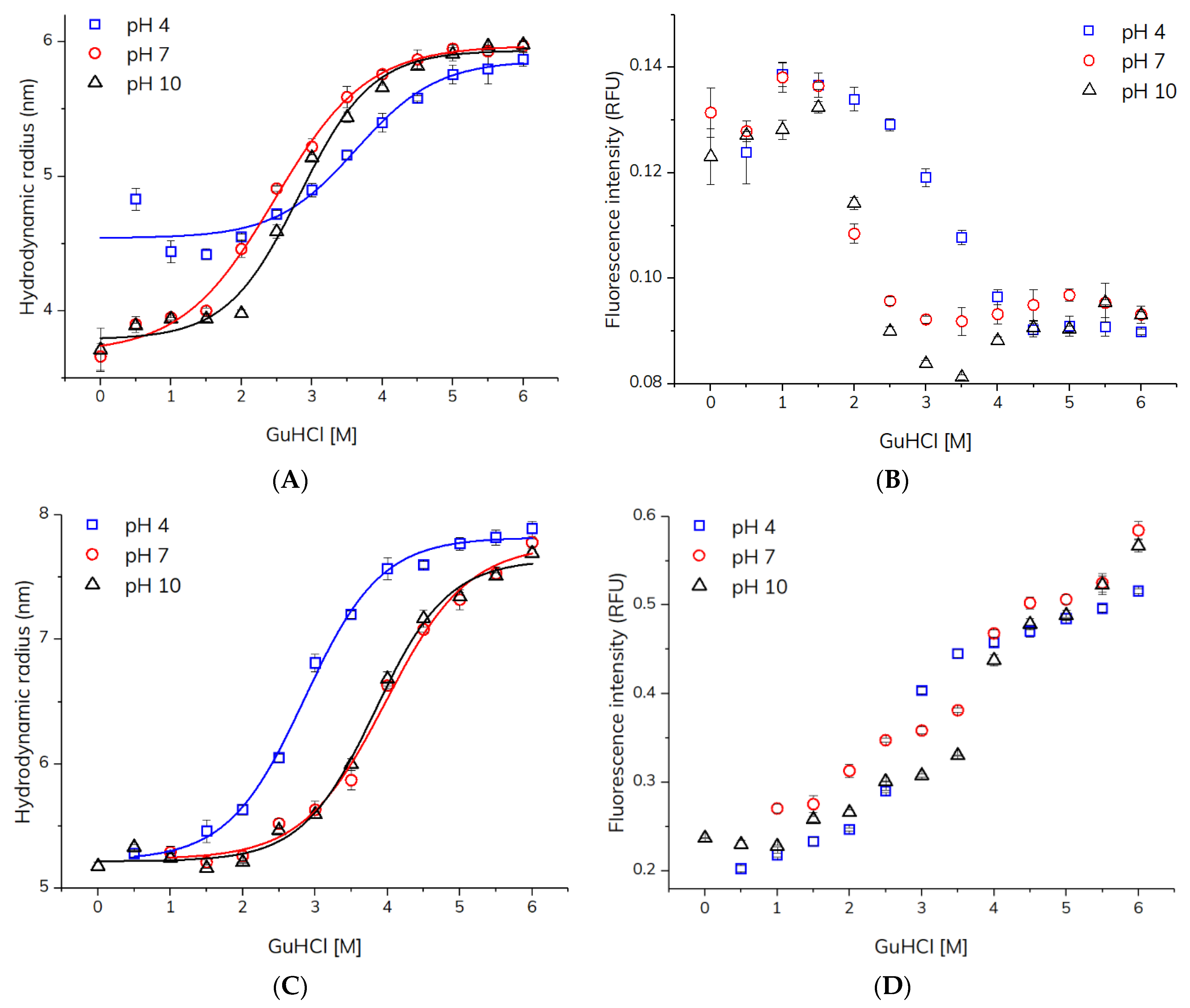
2.2. Unfolding of HSA and Adalimumab Using Capillary Mixing Mode
2.3. Quantitative Comparison of Stability towards Unfolding
3. Methods
3.1. Materials and Chemicals
Raw Materials
3.2. Equipment
3.3. Methods
3.3.1. Procedure
3.3.2. Denaturation Curves
3.3.3. Unfolding Kinetics of HSA
3.3.4. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Filipe, V.; Hawe, A.; Carpenter, J.F.; Jiskoot, W. Analytical approaches to assess the degradation of therapeutic proteins. TrAC Trends Anal. Chem. 2013, 49, 118–125. [Google Scholar] [CrossRef]
- Robinson, C.J.; Jones, C. Quality control and analytical techniques for biopharmaceuticals. Bioanalysis 2011, 3, 81–95. [Google Scholar] [CrossRef] [PubMed]
- US Food and Drug Administration. Points to Consider in the Manufacture and Testing of Monoclonal Antibody Products for Human Use. J. Immunother. 1997, 20, 214–215. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.M. Differential scanning calorimetry as a tool for protein folding and stability. Arch. Biochem. Biophys. 2013, 531, 100–109. [Google Scholar] [CrossRef]
- Leggio, C.; Galantini, L.; Konarev, P.V.; Pavel, N.V. Urea-Induced Denaturation Process on Defatted Human Serum Albumin and in the Presence of Palmitic Acid. J. Phys. Chem. B 2009, 113, 12590–12602. [Google Scholar] [CrossRef]
- Shriver, J.W. Protein Structure, Stability, and Interactions. Methods in Molecular Biology; Humana Press: Totowa, NJ, USA; Springer: Berlin/Heidelberg, Germany, 2009. [Google Scholar] [CrossRef] [Green Version]
- Hamborg, L.; Horsted, E.W.; Johansson, K.E.; Willemoës, M.; Lindorff-Larsen, K.; Teilum, K. Global analysis of protein stability by temperature and chemical denaturation. Anal. Biochem. 2020, 605, 113863. [Google Scholar] [CrossRef]
- Patra, M.; Mukhopadhyay, C.; Chakrabarti, A. Probing Conformational Stability and Dynamics of Erythroid and Nonerythroid Spectrin: Effects of Urea and Guanidine Hydrochloride. PLoS ONE 2015, 10, e0116991. [Google Scholar] [CrossRef]
- Jensen, H.; Østergaard, J. Flow Induced Dispersion Analysis Quantifies Noncovalent Interactions in Nanoliter Samples. J. Am. Chem. Soc. 2010, 132, 4070–4071. [Google Scholar] [CrossRef]
- Pedersen, M.E.; Haegebaert, R.M.S.; Østergaard, J.; Jensen, H. Size-based characterization of adalimumab and TNF-α interactions using flow induced dispersion analysis: Assessment of avidity-stabilized multiple bound species. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef]
- Pedersen, M.E.; Østergaard, J.; Jensen, H. In-Solution IgG Titer Determination in Fermentation Broth Using Affibodies and Flow-Induced Dispersion Analysis. ACS Omega 2020, 5, 10519–10524. [Google Scholar] [CrossRef]
- Poulsen, N.N.; Andersen, N.Z.; Østergaard, J.; Zhuang, G.; Petersen, N.J.; Jensen, H. Flow induced dispersion analysis rapidly quantifies proteins in human plasma samples. Analyst 2015, 140, 4365–4369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, G.I. Dispersion of soluble matter in solvent flowing slowly through a tube. Proc. R. Soc. Lond. Ser. A Math. Phys. Sci. 1953, 219, 186–203. [Google Scholar] [CrossRef]
- Ye, F.; Jensen, H.; Larsen, S.; Yaghmur, A.; Larsen, C.; Østergaard, J. Measurement of drug diffusivities in pharmaceutical solvents using Taylor dispersion analysis. J. Pharm. Biomed. Anal. 2012, 61, 176–183. [Google Scholar] [CrossRef]
- Cottet, H.; Biron, A.J.-P.; Martin, M. Taylor Dispersion Analysis of Mixtures. Anal. Chem. 2007, 79, 9066–9073. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.E.; Gad, S.I.; Østergaard, J.; Jensen, H. Protein Characterization in 3D: Size, Folding, and Functional Assessment in a Unified Approach. Anal. Chem. 2019, 91, 4975–4979. [Google Scholar] [CrossRef]
- Brahma, A.; Mandal, C.; Bhattacharyya, D. Characterization of a dimeric unfolding intermediate of bovine serum albumin under mildly acidic condition. Biochim. Biophys. Acta (BBA)-Proteins Proteom. 2005, 1751, 159–169. [Google Scholar] [CrossRef]
- Peters, T., Jr. All about Albumin: Biochemistry, Genetics, and Medical Applications; Academic Press: Cambridge, MA, USA, 1996. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, S.; Yamamoto, K.; Muniz, R.; Iwura, T. Physicochemical analysis and biological characterization of FKB327 as a biosimilar to adalimumab. Pharmacol. Res. Perspect. 2020, 8, e00604. [Google Scholar] [CrossRef]
- Di Stasio, E.; Bizzarri, P.; Misiti, F.; Pavoni, E.; Brancaccio, A. A fast and accurate procedure to collect and analyze unfolding fluorescence signal: The case of dystroglycan domains. Biophys. Chem. 2004, 107, 197–211. [Google Scholar] [CrossRef]
- Hawe, A.; Hulse, W.L.; Jiskoot, W.; Forbes, R.T. Taylor Dispersion Analysis Compared to Dynamic Light Scattering for the Size Analysis of Therapeutic Peptides and Proteins and Their Aggregates. Pharm. Res. 2011, 28, 2302–2310. [Google Scholar] [CrossRef] [Green Version]
- Krishnakumar, S.S.; Panda, D. Spatial Relationship between the Prodan Site, Trp-214, and Cys-34 Residues in Human Serum Albumin and Loss of Structure through Incremental Unfolding. Biochemistry 2002, 41, 7443–7452. [Google Scholar] [CrossRef]
- Ahmad, B.; Alam Khan, M.K.; Haq, S.K.; Khan, R.H. Intermediate formation at lower urea concentration in ‘B’ isomer of human serum albumin: A case study using domain specific ligands. Biochem. Biophys. Res. Commun. 2004, 314, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Schön, A.; Clarkson, B.R.; Siles, R.; Ross, P.; Brown, R.K.; Freire, E. Denatured state aggregation parameters derived from concentration dependence of protein stability. Anal. Biochem. 2015, 488, 45–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farruggia, B.; Picó, G.A. Thermodynamic features of the chemical and thermal denaturations of human serum albumin. Int. J. Biol. Macromol. 1999, 26, 317–323. [Google Scholar] [CrossRef]
- Chamieh, J.; Merdassi, H.; Rossi, J.C.; Jannin, V.; Demarne, F.; Cottet, H. Size Characterization of Lipid-Based Self-Emulsifying Pharmaceutical Excipients during Lipolysis Using Taylor Dispersion Analysis with Fluorescence Detection. Int. J. Pharm. 2018, 537, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Latunde-Dada, S.; Bott, R.; Hampton, K.; Patel, J.; Leszczyszyn, O.I. Methodologies for the Taylor Dispersion Analysis for Mixtures, Aggregates and the Mitigation of Buffer Mismatch Effects. Anal. Methods 2015, 7, 10312–10321. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| pH | Adalimumab | HAS |
|---|---|---|
| Cm/M [GuHCl] | ||
| 4.0 | 2.6 | 3.5 |
| 7.0 | 3.8 | 2.2 |
| 10.0 | 3.6 | 2.6 |
| pH | ΔG° (H2O) (kcal/mol) | |
| 4.0 | 3.0 | 3.6 |
| 7.0 | 3.9 | 2.0 |
| 10.0 | 4.3 | 3.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pedersen, M.E.; Østergaard, J.; Jensen, H. Quantification of Structural Integrity and Stability Using Nanograms of Protein by Flow-Induced Dispersion Analysis. Molecules 2022, 27, 2506. https://doi.org/10.3390/molecules27082506
Pedersen ME, Østergaard J, Jensen H. Quantification of Structural Integrity and Stability Using Nanograms of Protein by Flow-Induced Dispersion Analysis. Molecules. 2022; 27(8):2506. https://doi.org/10.3390/molecules27082506
Chicago/Turabian StylePedersen, Morten E., Jesper Østergaard, and Henrik Jensen. 2022. "Quantification of Structural Integrity and Stability Using Nanograms of Protein by Flow-Induced Dispersion Analysis" Molecules 27, no. 8: 2506. https://doi.org/10.3390/molecules27082506
APA StylePedersen, M. E., Østergaard, J., & Jensen, H. (2022). Quantification of Structural Integrity and Stability Using Nanograms of Protein by Flow-Induced Dispersion Analysis. Molecules, 27(8), 2506. https://doi.org/10.3390/molecules27082506