
Revisiting the Synthesis of Functionally Substituted 1,4-Dihydrobenzo[e][1,2,4]triazines


Abstract
:1. Introduction
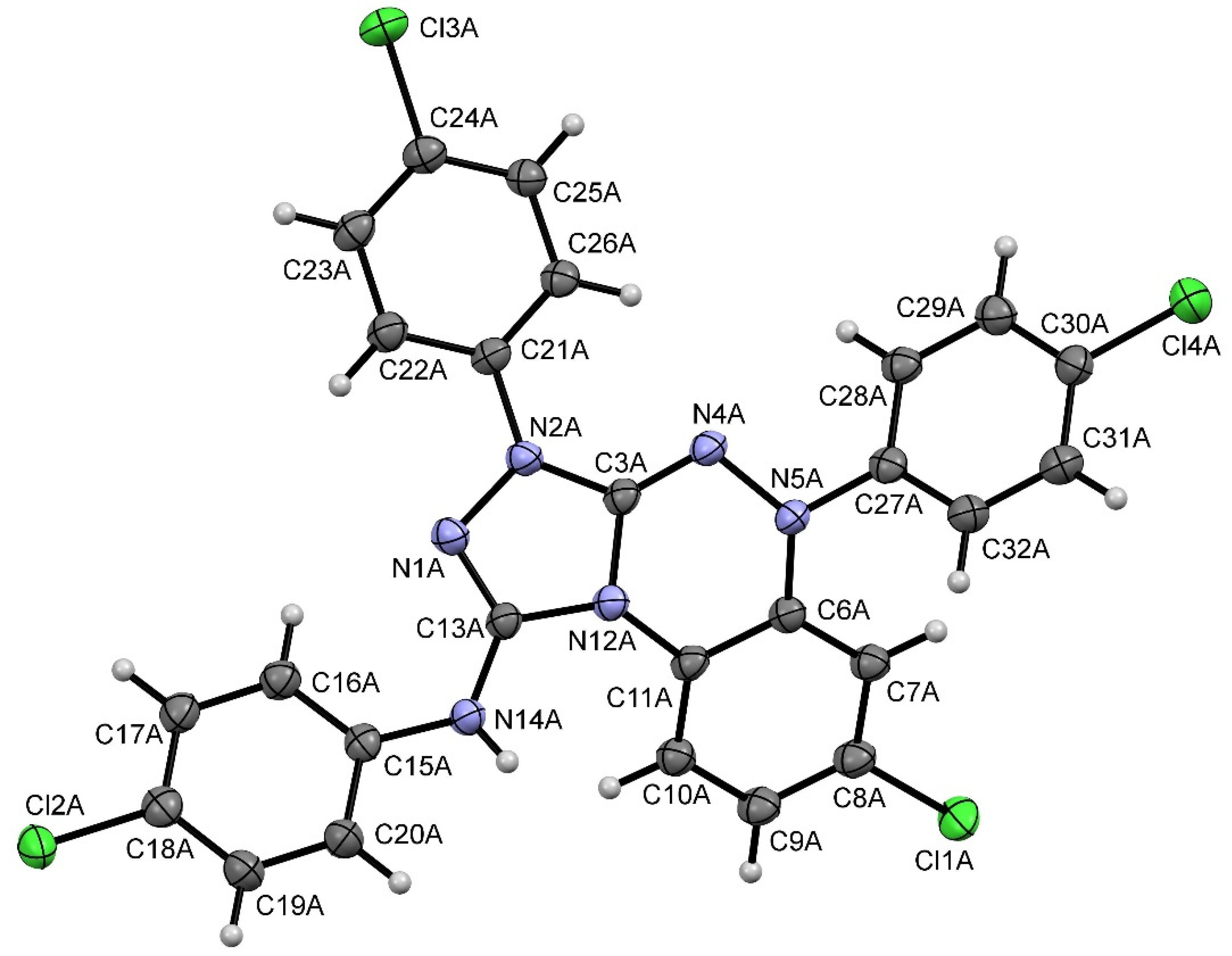
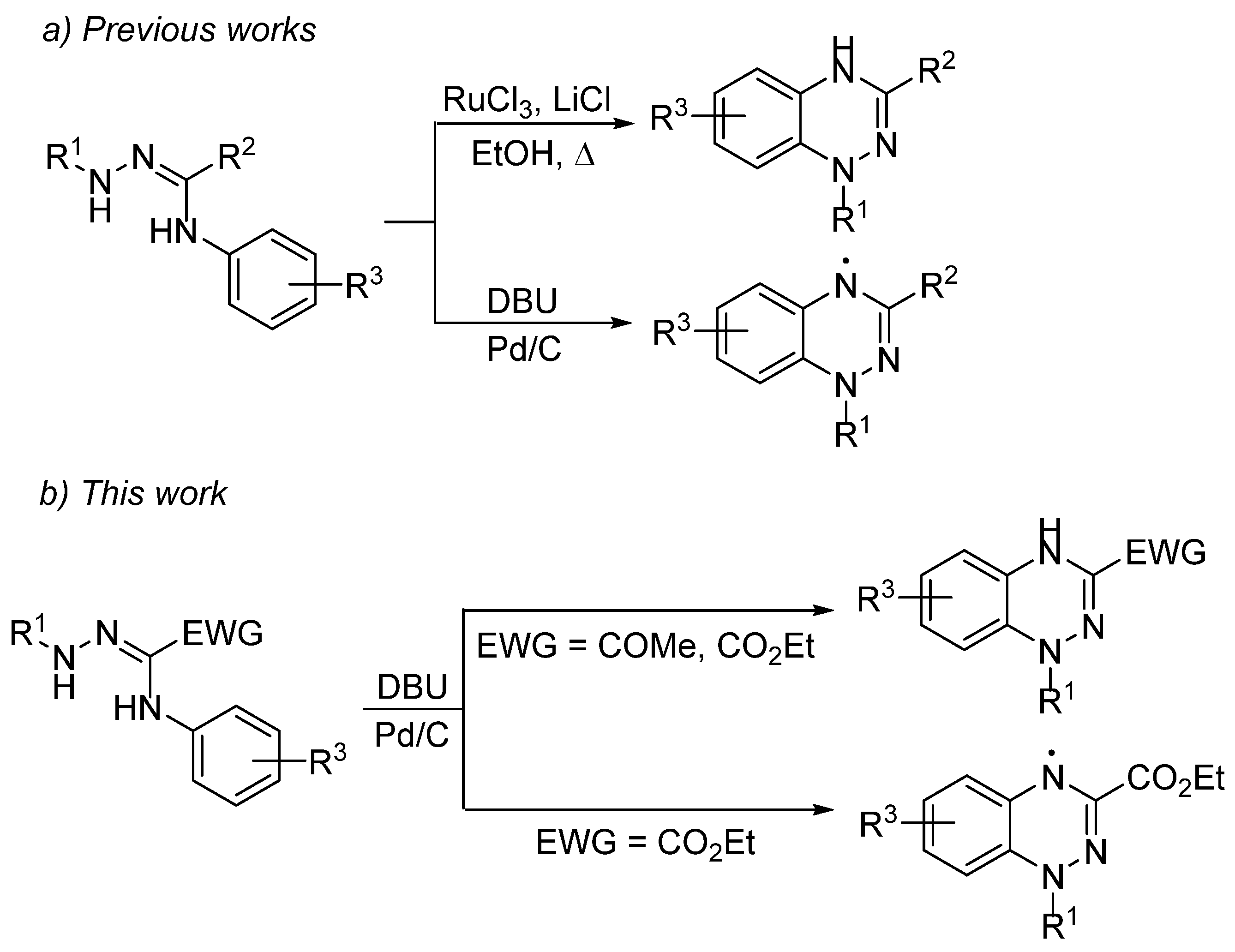
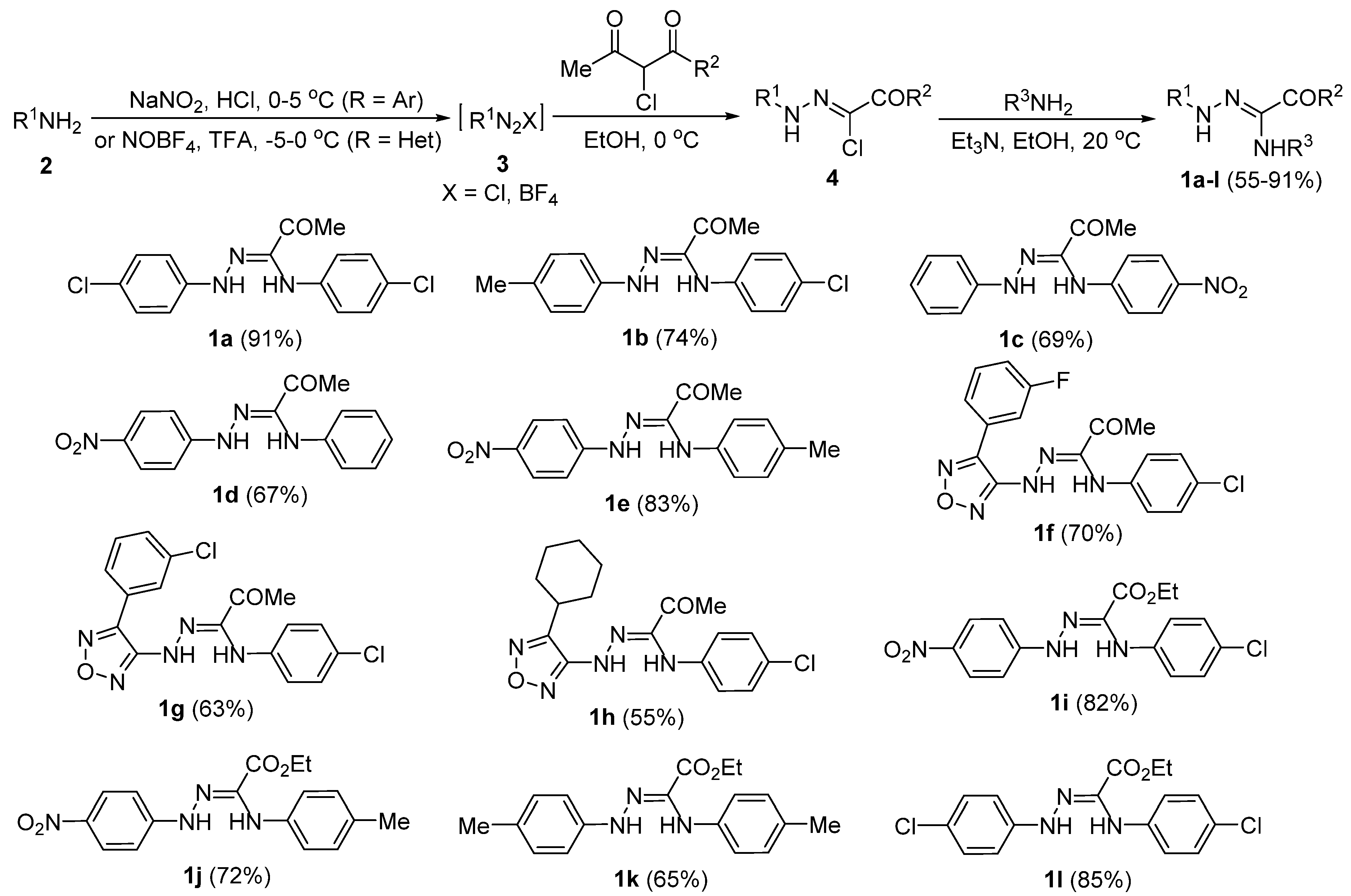
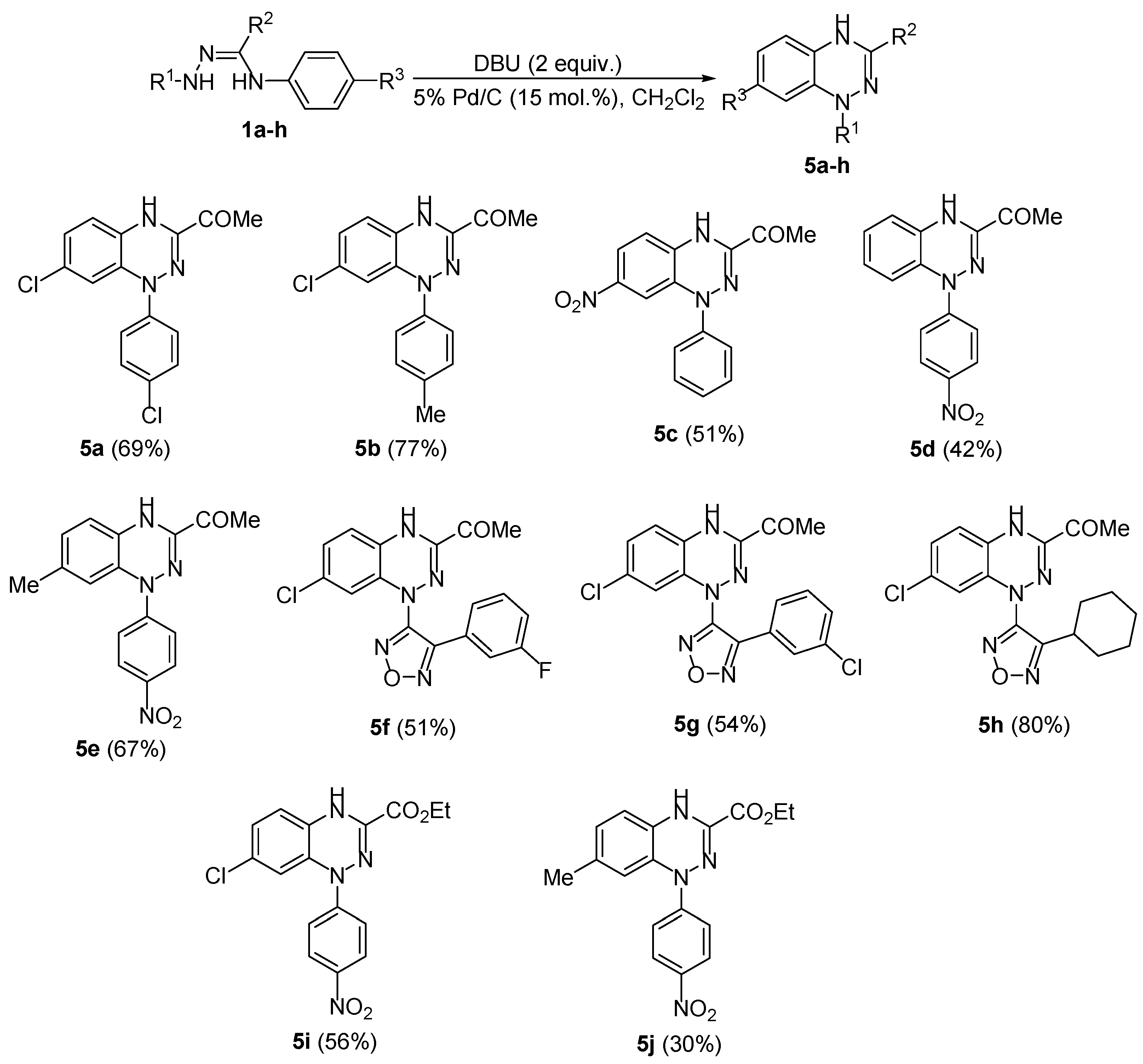
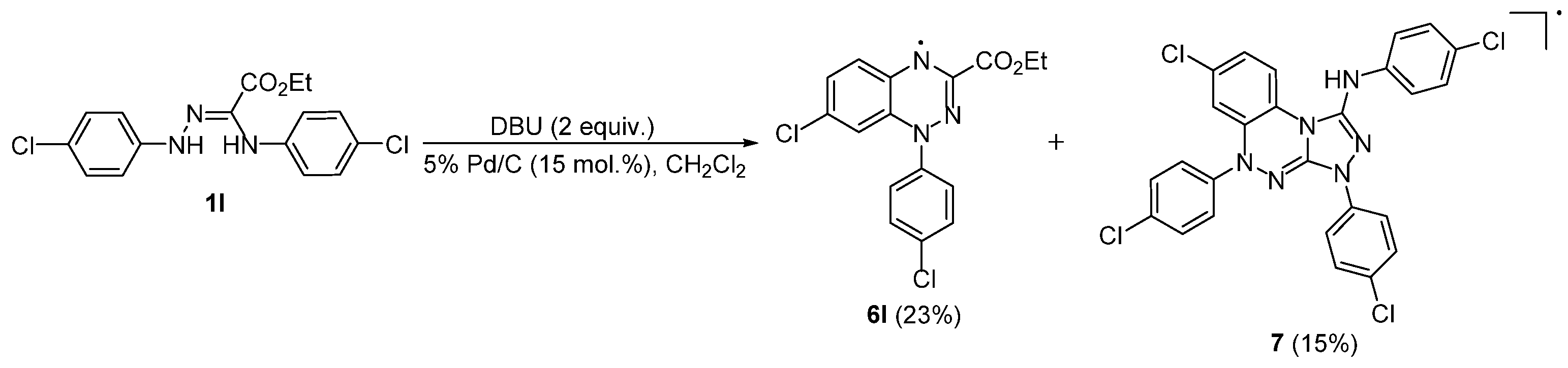
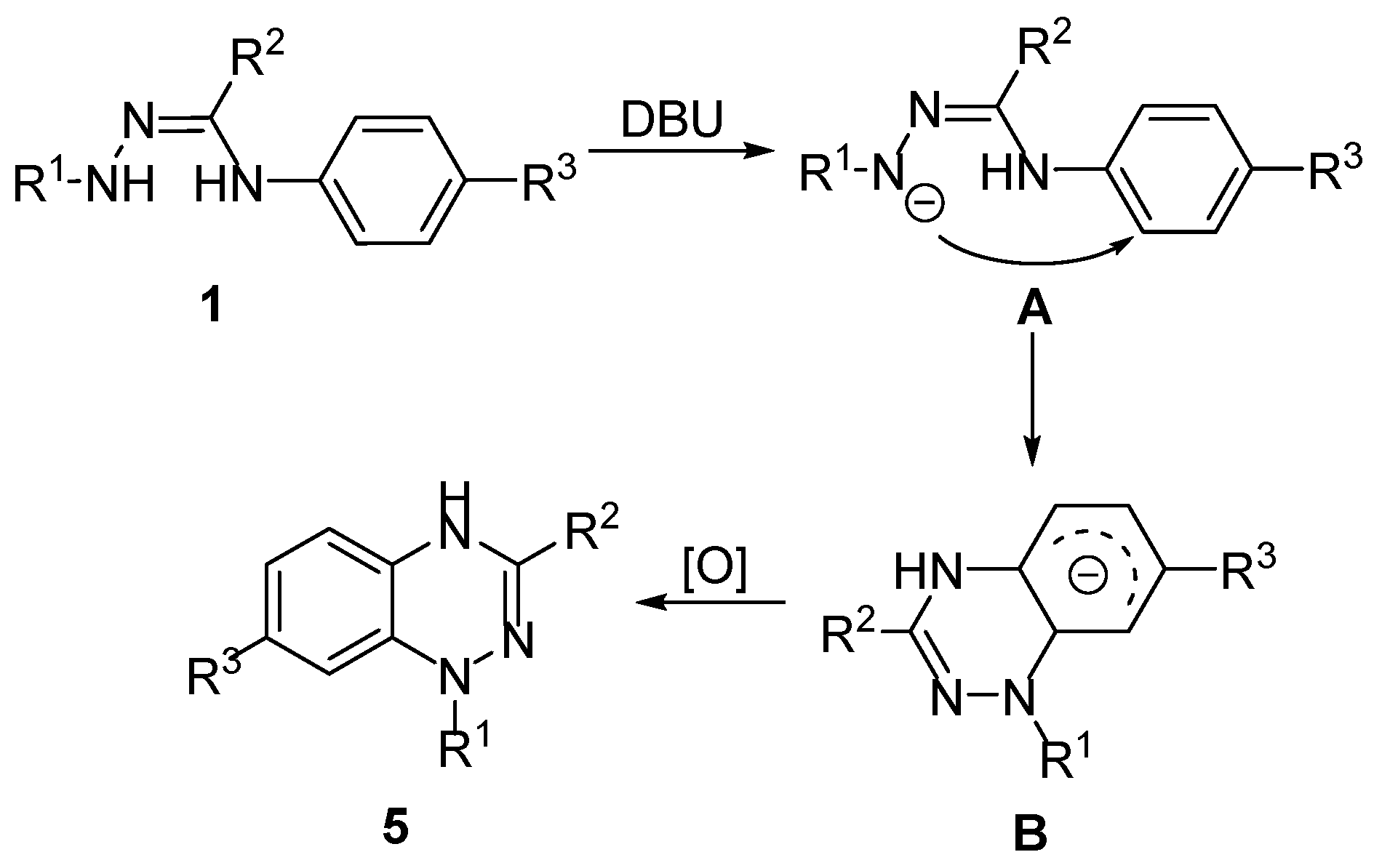
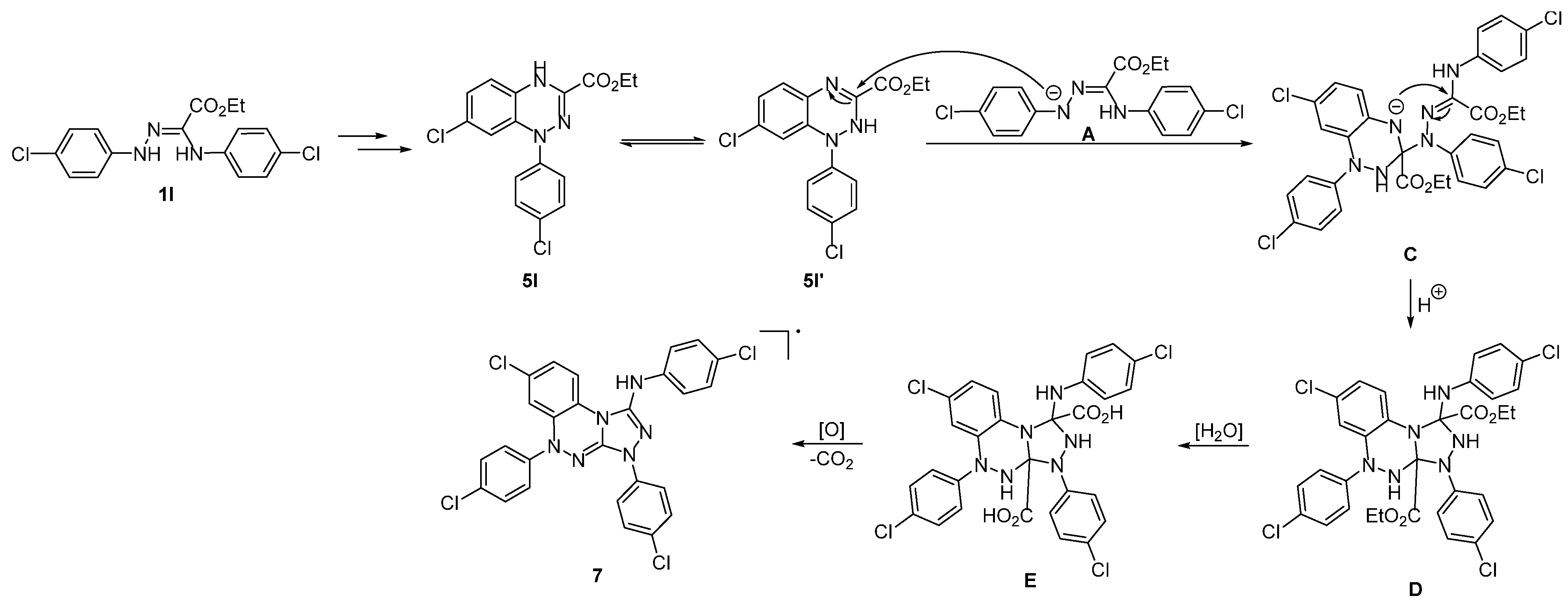
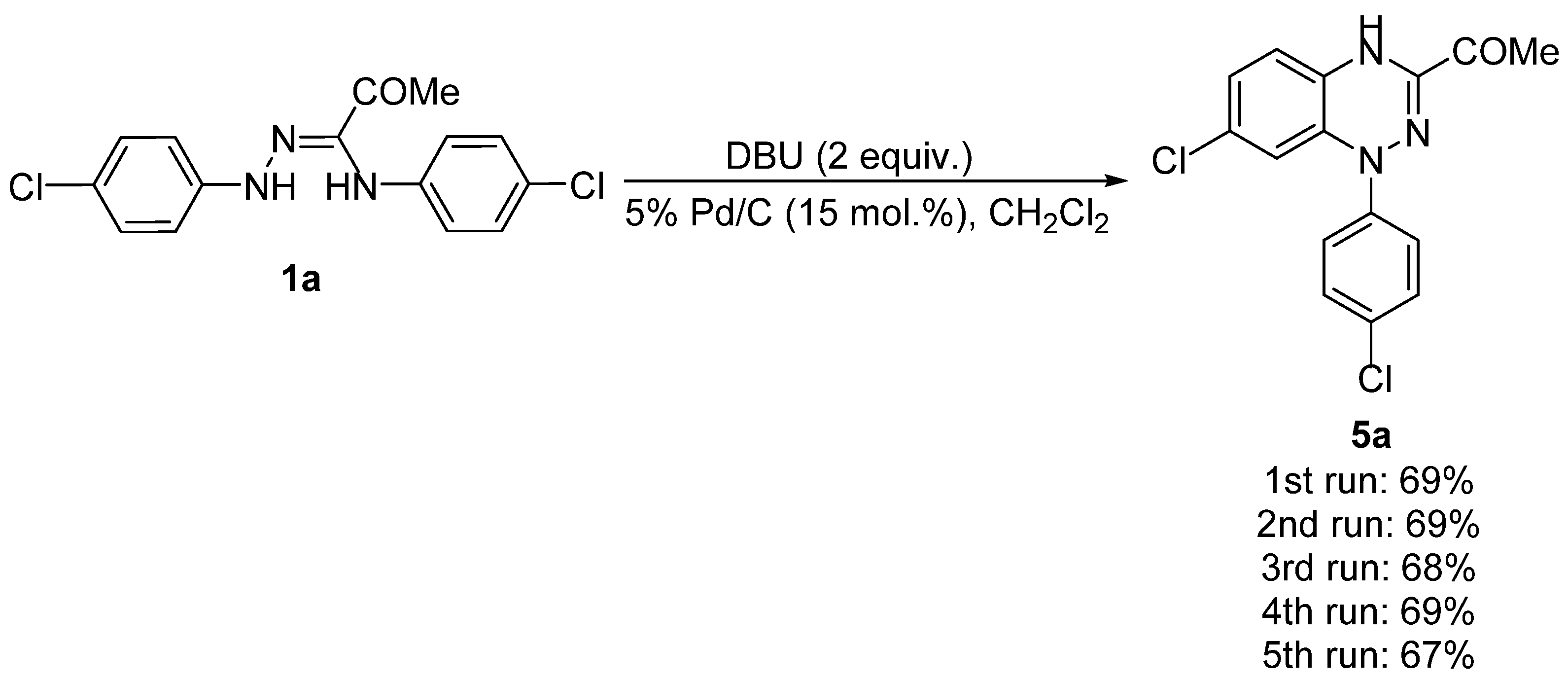
2. Results and Discussion
3. Conclusions
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Li, D.; Yu, G. Innovation of Materials, Devices, and Functionalized Interfaces in Organic Spintronics. Adv. Funct. Mater. 2021, 31, 2100550. [Google Scholar] [CrossRef]
- Liu, C.-Y.; Lin, P.-H.; Lee, K.-M. Development of Step-Saving Alternative Synthetic Pathways for Functional π-Conjugated Materials. Chem. Rec. 2021, 21, 3498–3508. [Google Scholar] [CrossRef] [PubMed]
- Sathiyan, G.; Wang, H.; Chen, C.; Miao, Y.; Zhai, M.; Cheng, M. Impact of fluorine substitution in organic functional materials for perovskite solar cell. Dyes Pigm. 2022, 198, 110029. [Google Scholar] [CrossRef]
- Yang, X.-D.; Tan, L.; Sun, J.-K. Encapsulation of Metal Clusters within Porous Organic Materials: From Synthesis to Catalysis Applications. Chem. Asian J. 2022, 17, e202101289. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Das, S.K.; Khatua, H.; Das, S.; Chattopadhyay, B. Road Map for the Construction of High-Valued N-Heterocycles via Denitrogenative Annulation. Acc. Chem. Res. 2021, 54, 4395–4409. [Google Scholar] [CrossRef] [PubMed]
- Odom, A.L.; McDaniel, T.J. Titanium-Catalyzed Multicomponent Couplings: Efficient One-Pot Syntheses of Nitrogen Heterocycles. Acc. Chem. Res. 2015, 48, 2822–2833. [Google Scholar] [CrossRef] [PubMed]
- Makhova, N.N.; Belen’kii, L.I.; Gazieva, G.A.; Dalinger, I.L.; Konstantinova, L.S.; Kuznetsov, V.V.; Kravchenko, A.N.; Krayushkin, M.M.; Rakitin, O.A.; Starosotnikov, A.M.; et al. Progress in the chemistry of nitrogen-, oxygen- and sulfur-containing heterocyclic systems. Russ. Chem. Rev. 2020, 89, 55–124. [Google Scholar] [CrossRef]
- Ziarani, G.M.; Mostofi, M.; Gholamzadeh, P.; Mohammadi-Khanaposhtani, M.; Yavari, H. The synthesis of 1,2,4-benzotriazines. Arkivoc 2019, i, 41–105. [Google Scholar] [CrossRef] [Green Version]
- Mahran, A.M.; Farghaly, T.A.; Nada, A.A. Hydrazonoyl halides in heterocycles: Synthesis and anti-microbial activity of new 1,2,4-benzotriazine and bis-1,2,4-benzotriazine derivatives. Res. Chem. Intermed. 2015, 41, 2961–2969. [Google Scholar] [CrossRef]
- Keivanloo, A.; Bakherad, M.; Lotfi, M. Use of ligand-assisted click reactions for the rapid synthesis of novel 1,2,3-triazole pharmacophore-based 1,2,4-triazines and their benzofused analogues. Tetrahedron 2017, 73, 5872–5882. [Google Scholar] [CrossRef]
- Ji, Y.; Long, L.; Zheng, Y. Recent advances of stable Blatter radicals: Synthesis, properties and applications. Mater. Chem. Front. 2020, 4, 3433–3443. [Google Scholar] [CrossRef]
- Rogers, F.J.M.; Norcott, P.L.; Coote, M.L. Recent advances in the chemistry of benzo[e][1,2,4]triazinyl radicals. Org. Biomol. Chem. 2020, 18, 8255–8277. [Google Scholar] [CrossRef] [PubMed]
- Constantinides, C.P.; Koutentis, P.A. Stable N- and N/S-Rich Heterocyclic Radicals: Synthesis and Applications. Adv. Heterocycl. Chem. 2016, 119, 173–207. [Google Scholar] [CrossRef]
- Sidharth, T.N.S.; Nasani, R.; Gupta, A.; Sooraj, B.N.S.; Roy, S.; Mondal, A.; Konar, S. Reversal of magnetic exchange coupling between copper(II) and Blatter radical depending on the coordination environment. Inorg. Chim. Acta 2020, 503, 119395. [Google Scholar] [CrossRef]
- Kapuściński, S.; Szczytko, J.; Pociecha, D.; Jasiński, M.; Kaszyński, P. Discs, dumbbells and superdiscs: Molecular and supermolecular architecture dependent magnetic behavior of mesogenic Blatter radical derivatives. Mater. Chem. Front. 2021, 5, 6512–6521. [Google Scholar] [CrossRef]
- Constantinides, C.P.; Lawson, D.B.; Zissimou, G.A.; Berezin, A.A.; Mailman, A.; Manoli, M.; Kourtellaris, A.; Leitus, G.M.; Clérac, R.; Tuononen, H.M.; et al. Polymorphism in a π stacked Blatter radical: Structures and magnetic properties of 3-(phenyl)-1-(pyrid-2-yl)-1,4-dihydrobenzo[e][1,2,4]triazin-4-yl. CrystEngComm 2020, 22, 5453–5463. [Google Scholar] [CrossRef]
- Constantinides, C.P.; Lawson, D.B.; Berezin, A.A.; Zissimou, G.A.; Manoli, M.; Leitus, G.M.; Koutentis, P.A. Ferromagnetic interactions in a 1D Heisenberg linear chain of 1-phenyl-3,7-bis(trifluoromethyl)-1,4-dihydro-1,2,4-benzotriazin-4-yls. CrystEngComm 2019, 21, 4599–4606. [Google Scholar] [CrossRef]
- Kapuściński, S.; Anand, B.; Bartos, P.; Garcia Fernandez, J.M.; Kaszyński, P. Tethered Blatter Radical for Molecular Grafting: Synthesis of 6-Hydroxyhexyloxy, Hydroxymethyl, and Bis(hydroxymethyl) Derivatives and Their Functionalization. Molecules 2022, 27, 1176. [Google Scholar] [CrossRef]
- Kaszyński, P.; Kapuściński, S.; Ciastek-Iskrzycka, S. Liquid crystalline derivatives of heterocyclic radicals. Adv. Heterocycl. Chem. 2019, 128, 263–331. [Google Scholar] [CrossRef]
- Jasiński, M.; Szczytko, J.; Pociecha, D.; Monobe, H.; Kaszyński, P. Substituent-Dependent Magnetic Behavior of Discotic Benzo[e][1,2,4]triazinyls. J. Am. Chem. Soc. 2016, 138, 9421–9424. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, Y.; Zhou, H.; Miao, M.S.; Wudl, F.; Nguyen, T.Q. Temperature Tunable Self-Doping in Stable Diradicaloid Thin-Film Devices. Adv. Mater. 2015, 27, 7412–7419. [Google Scholar] [CrossRef] [PubMed]
- Calzolari, A.; Rajca, A.; Casu, M.B. From radical to triradical thin film processes: The Blatter radical derivatives. J. Mater. Chem. C 2021, 9, 10787–10793. [Google Scholar] [CrossRef]
- Morgan, I.S.; Mansikkamäki, A.; Zissimou, G.A.; Koutentis, P.A.; Rouzières, M.; Clérac, R.; Tuononen, H.M. Coordination Complexes of a Neutral 1,2,4-Benzotriazinyl Radical Ligand: Synthesis, Molecular and Electronic Structures, and Magnetic Properties. Chem. Eur. J. 2015, 21, 15843–15853. [Google Scholar] [CrossRef]
- Karecla, G.; Papagiorgis, P.; Panagi, N.; Zissimou, G.A.; Constantinides, C.P.; Koutentis, P.A.; Itskos, G.; Hayes, S.C. Emission from the stable Blatter radical. New J. Chem. 2017, 41, 8604–8613. [Google Scholar] [CrossRef]
- Zissimou, G.A.; Berezin, A.A.; Manoli, M.; Nicolaides, C.; Trypiniotis, T.; Koutentis, P.A. 3,3′,3′-(Benzene-1,3,5-triyl)tris(1-phenyl-1H-benzo[e][1,2,4]triazin-4-yl): A C3 symmetrical Blatter-type triradical. Tetrahedron 2020, 76, 131077. [Google Scholar] [CrossRef]
- Al-Noaimi, M.Z.; Abdel-Jalil, R.J.; El-Abadelah, M.M.; Haddad, S.F.; Baqi, Y.N.H.; Voelter, W. Metal-Assisted Oxidative Cyclization of Arylamidrazones I. Synthesis of 3-Acetyl-1,4-dihydro-1-phenyl-1,2,4-benzotriazine. Monat. Chem. 2006, 137, 745–750. [Google Scholar] [CrossRef]
- Koutentis, P.A.; Lo Re, D. Catalytic Oxidation of N-Phenylamidrazones to 1,3-Diphenyl-1,4-dihydro-1,2,4-benzotriazin-4-yls: An Improved Synthesis of Blatter’s Radical. Synthesis 2010, 2010, 2075–2079. [Google Scholar] [CrossRef]
- Bodzioch, A.; Zheng, M.; Kaszyński, P.; Utecht, G. Functional Group Transformations in Derivatives of 1,4-Dihydrobenzo[1,2,4]triazinyl Radical. J. Org. Chem. 2014, 79, 7294–7310. [Google Scholar] [CrossRef]
- Ciccullo, F.; Gallagher, N.M.; Geladari, O.; Chassé, T.; Rajca, A.; Casu, M.B. A Derivative of the Blatter Radical as a Potential Metal-Free Magnet for Stable Thin Films and Interfaces. ACS Appl. Mater. Interfaces 2016, 8, 1805–1812. [Google Scholar] [CrossRef]
- Titenkova, K.Y.; Shaferov, A.V.; Larin, A.A.; Epishina, M.A.; Kulikov, A.S.; Ananyev, I.V.; Makhova, N.N. Tandem acid-promoted intramolecular azide-hydrazone electrocyclization/hydrolysis approach for the synthesis of N-aminotetrazoles. Tetrahedron 2022, 103, 132563. [Google Scholar] [CrossRef]
- Arafeh, M.M.; Moghadam, E.S.; Adham, S.A.I.; Stoll, R.; Abdel-Jalil, R.J. Synthesis and Cytotoxic Activity Study of Novel 2-(Aryldiazenyl)-3-methyl-1H-benzo[g]indole Derivatives. Molecules 2021, 26, 4240. [Google Scholar] [CrossRef] [PubMed]
- CrysAlisPro. Version 1.171.41.106a. In Rigaku Oxford Diffraction; Rigaku Corporation: Oxford, UK, 2021. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 229–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. Mercury: Visualization and Analysis of Crystal Structures. J. Appl. Cryst. 2006, 39, 453–457. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
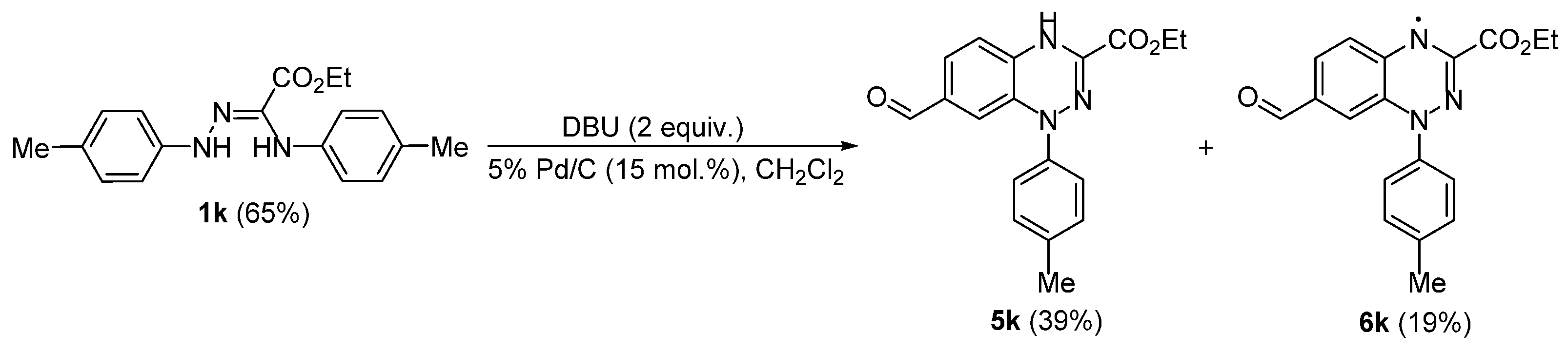{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Base (Equiv.) | Additive (mol.%) | Solvent | T, °C | Time, h | Product (Yield, %) b |
|---|---|---|---|---|---|---|
| 1 | DBU (0.1) | 5% Pd/C (1.6) | CH2Cl2 | 25 | 72 | - c |
| 2 | DBU (1) | - | CH2Cl2 | 25 | 72 | - c |
| 3 | DBU (1) | 5% Pd/C (15) | CH2Cl2 | 25 | 10 | 5a (43) |
| 4 | DBU (1) | 5% Pd/C (15) | CH2Cl2 | 40 | 7 | 5a (36) |
| 5 | DBU (2) | 5% Pd/C (15) | CH2Cl2 | 25 | 8 | 5a (69) |
| 6 | Et3N (2) | 5% Pd/C (15) | CH2Cl2 | 25 | 48 | 5a (29) |
| 7 | DBU (2) | Cact (200) | CH2Cl2 | 40 | 24 | 5a (35) |
| 8 | DBU (2) | TiO2 (50) | CH2Cl2 | 40 | 24 | 5a (8) |
| 9 | Cs2CO3 (1) | - | CH2Cl2 | 25 | 48 | 5a (10) |
| 10 | - | MnO2 (100) | CH2Cl2 | 25 | 72 | - c |
| 11 | DBU (2) | 5% Pd/C (15) | MeCN | 50 | 8 | 5a (23) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Epishina, M.A.; Kulikov, A.S.; Fershtat, L.L. Revisiting the Synthesis of Functionally Substituted 1,4-Dihydrobenzo[e][1,2,4]triazines. Molecules 2022, 27, 2575. https://doi.org/10.3390/molecules27082575
Epishina MA, Kulikov AS, Fershtat LL. Revisiting the Synthesis of Functionally Substituted 1,4-Dihydrobenzo[e][1,2,4]triazines. Molecules. 2022; 27(8):2575. https://doi.org/10.3390/molecules27082575
Chicago/Turabian StyleEpishina, Margarita A., Alexander S. Kulikov, and Leonid L. Fershtat. 2022. "Revisiting the Synthesis of Functionally Substituted 1,4-Dihydrobenzo[e][1,2,4]triazines" Molecules 27, no. 8: 2575. https://doi.org/10.3390/molecules27082575
APA StyleEpishina, M. A., Kulikov, A. S., & Fershtat, L. L. (2022). Revisiting the Synthesis of Functionally Substituted 1,4-Dihydrobenzo[e][1,2,4]triazines. Molecules, 27(8), 2575. https://doi.org/10.3390/molecules27082575