
Synthesis of Tetrapeptides Containing Dehydroalanine, Dehydrophenylalanine and Oxazole as Building Blocks for Construction of Foldamers and Bioinspired Catalysts

 , ,
, , 
Abstract
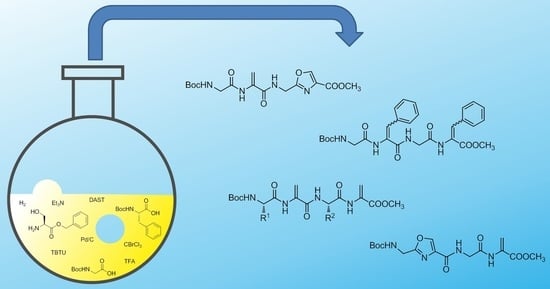
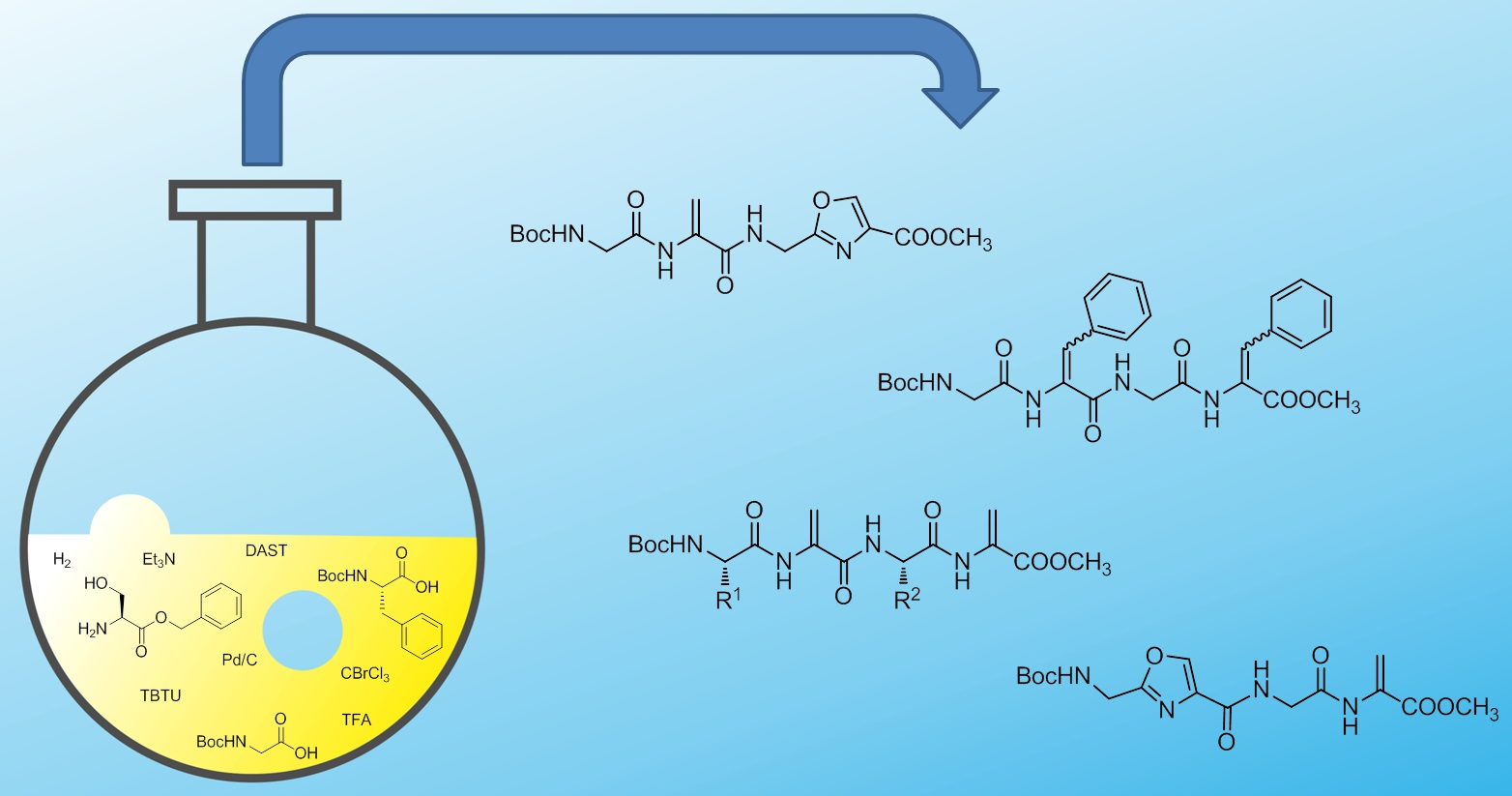
:
1. Introduction
2. Results
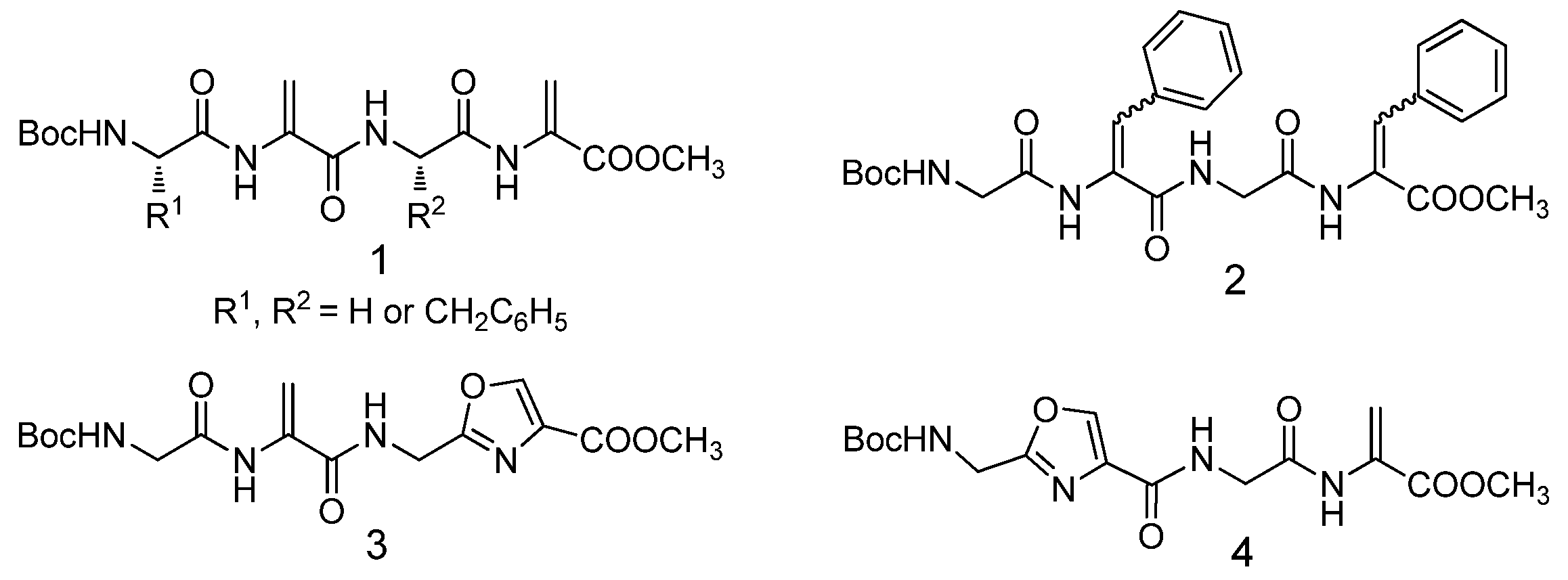
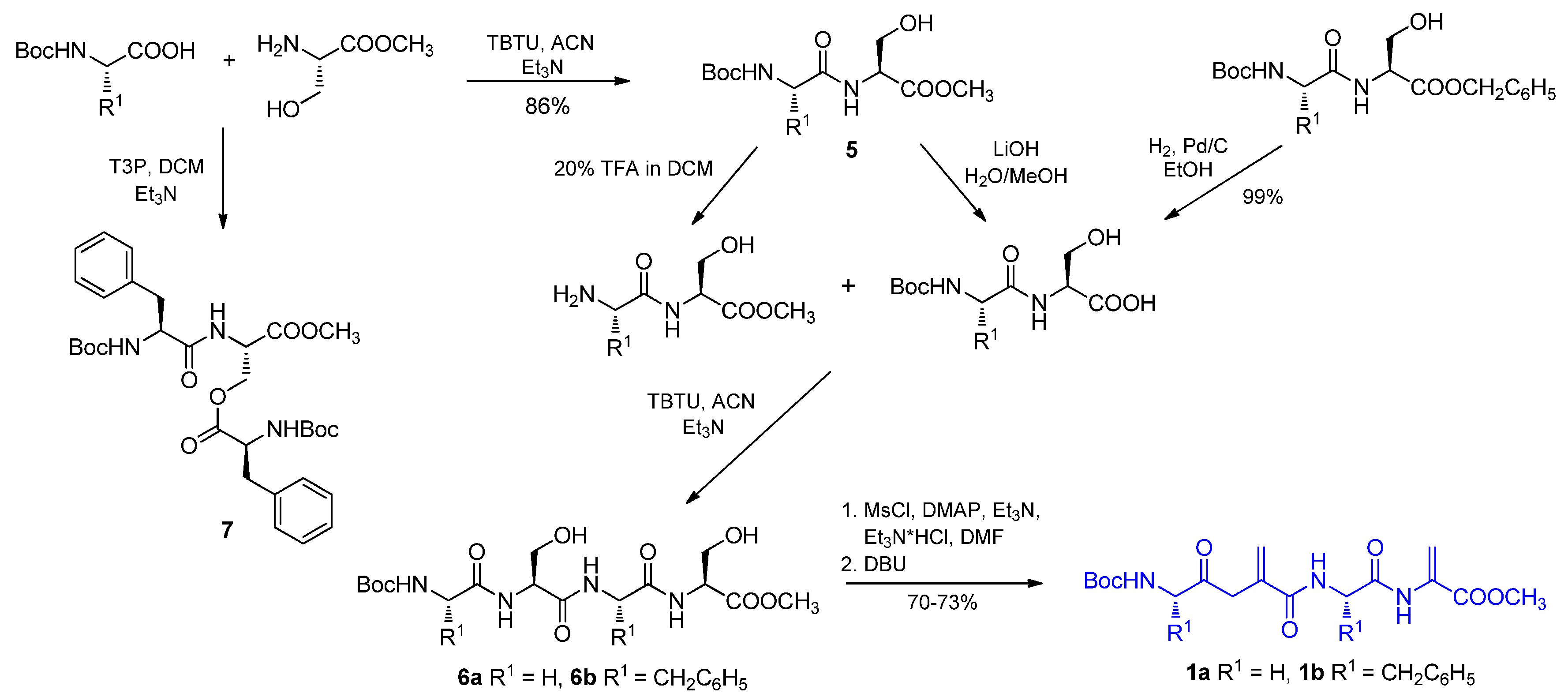
2.1. Tetrapeptides Containing Two Dehydroalanine Units
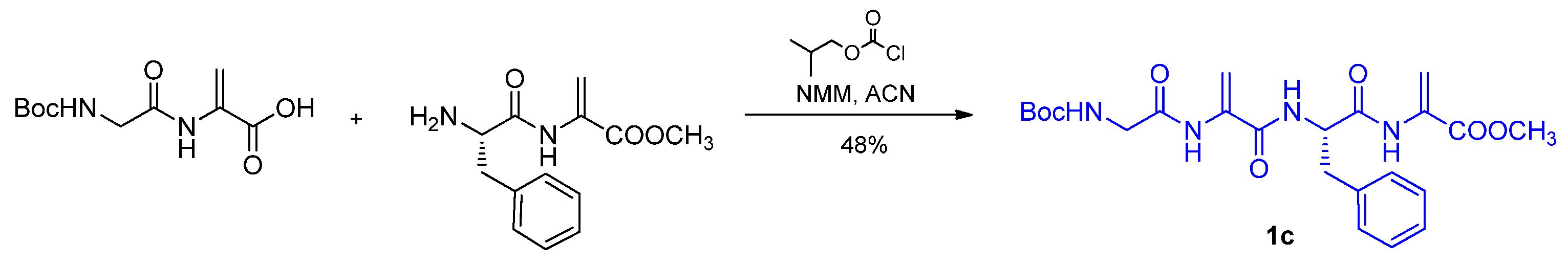
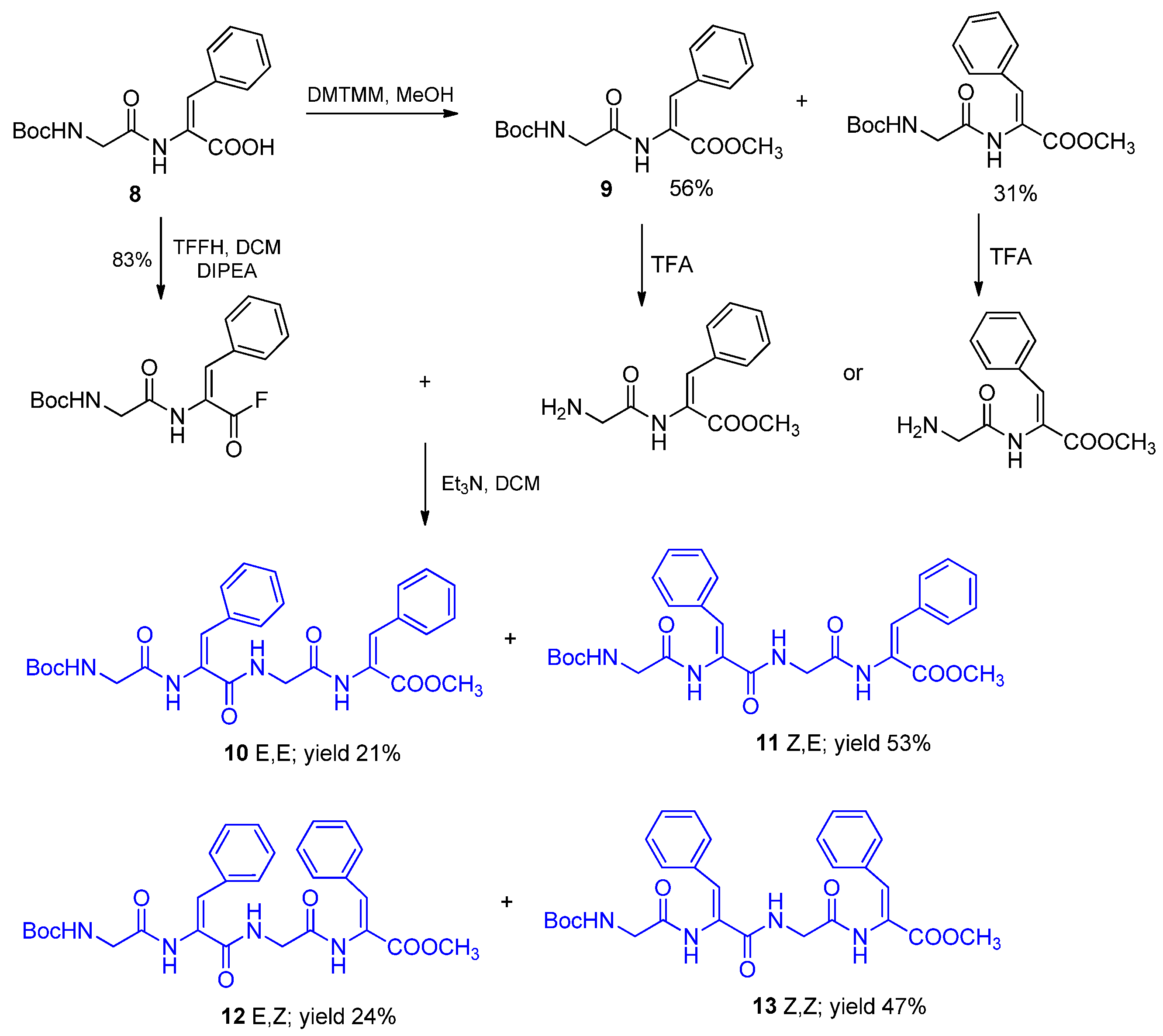
2.2. Tetrapeptides Containing Two Dehydrophenylalanine Units
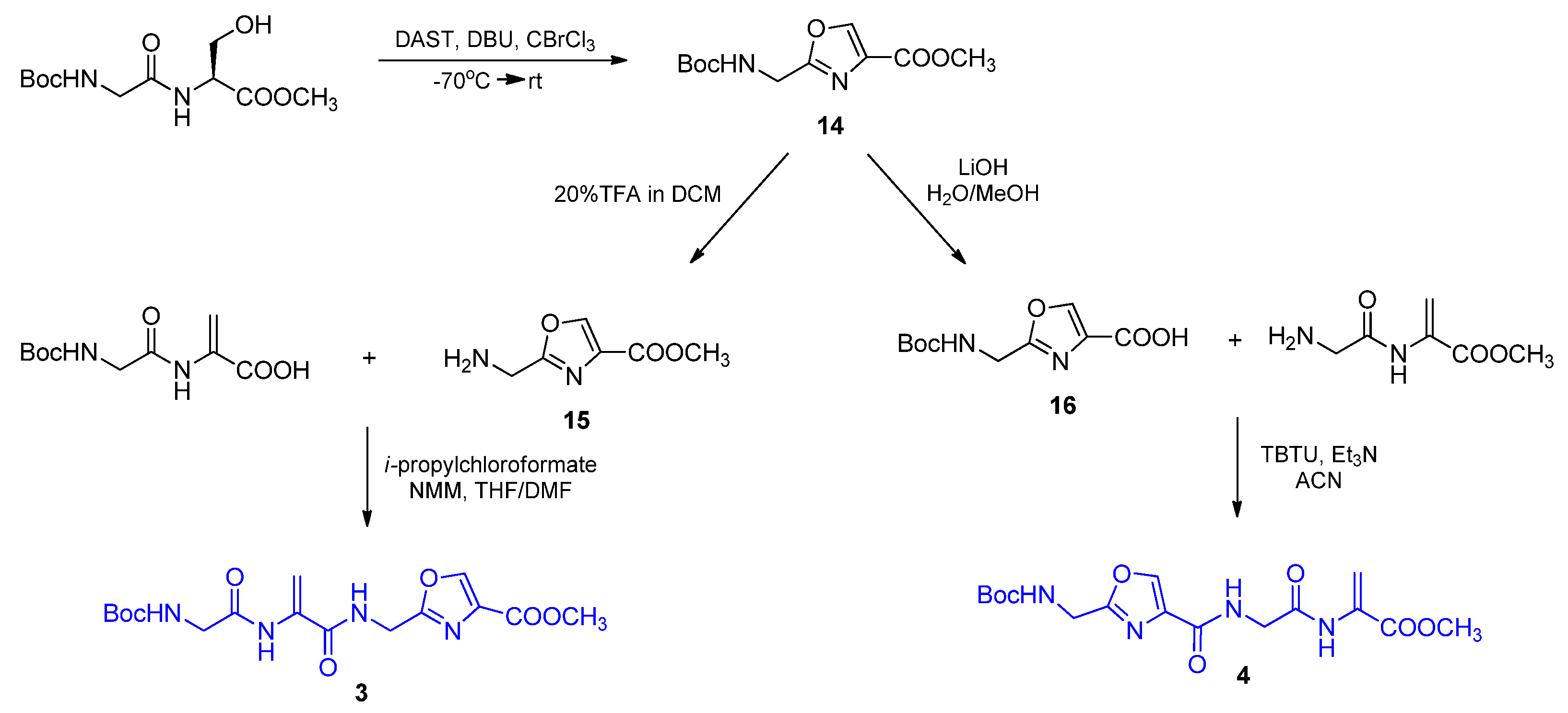
2.3. Hybrid Tetrapeptides Containing Dehydroalanine and Oxazole Units
3. Conclusions
4. Experimental Section
4.1. Materials and Methods
4.2. Syntheses
4.2.1. Substrates
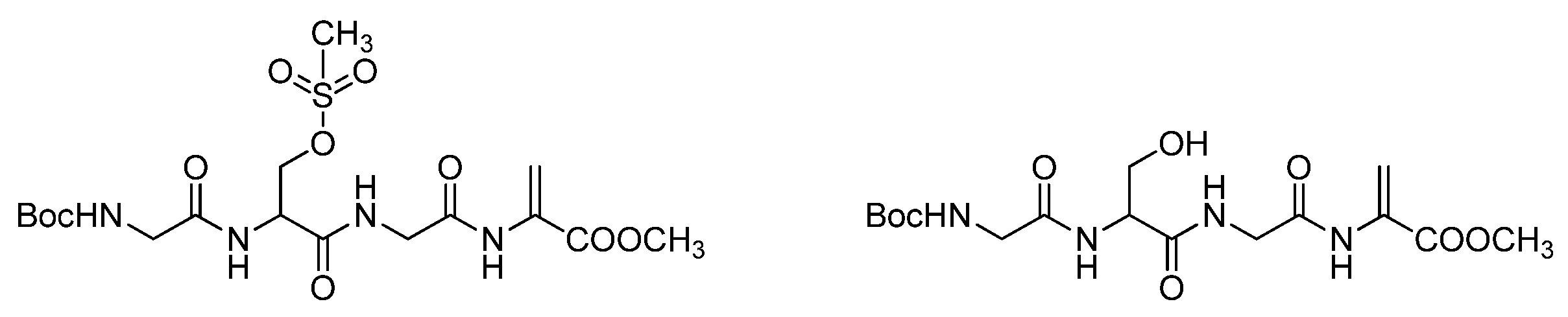
4.2.2. Dehydratation of Tetrapeptides 6a or 6b—An Optimized Procedure
- ▪
- Methyl Boc-glycyl-dehydroalanyl-glycyl-dehydroalaninate (Compound 1a)
- ▪
- Methyl Boc-l-phenylalanyl-dehydroalanyl-l-phenylalanyl-dehydroalaninate (Compound 1b)
4.2.3. Synthesis of Tetrapeptides and Their Oxazole Counterparts by Coupling Reaction
- ▪
- Methyl Boc-glycyl-dehydroalanyl-l-phenylalanyl-dehydroalaninate (Compound 1c)
- ▪
- Methyl 2-(1-(tert-butoxycarbonyl-glycyl-dehydroalanyl)methyl)oxazole-4-carboxylate (Compound 3)
- ▪
- Methyl 2-(1-(tert-butoxycarbonylamino)methyl)oxazole-4-glycyl-dehydroalaninate (Compound 4)
- ▪
- Methyl Boc-glycyl-l-serinyl-glycyl-l-serinate (Compound 6a)
- ▪
- Methyl Boc-l-phenylalanyl-l-serinyl-l-phenylalanyl-l-serinate (Compound 6b)
4.2.4. Isomers of Methyl Boc-glycyl-dehydrophenyalanyl-glycyl-dehydrophenyalanine (Compounds 10–13)
- Acylation of Gly-EΔPhe-OMe
- ▪
- Methyl Boc-glycyl-(E)-dehydrophenyalanyl-glycyl-(E)-dehydrophenyalanine (Compound 10)
- ▪
- Methyl Boc-glycyl-(Z)-dehydrophenyalanyl-glycyl-(E)-dehydrophenyalanine (Compound 11)
- Acylation of Gly-ZΔPhe-OMe
- ▪
- Methyl Boc-glycyl-(E)-dehydrophenyalanyl-glycyl-(Z)-dehydrophenyalanine (Compound 12)
- ▪
- Methyl Boc-glycyl-(Z)-dehydrophenyalanyl-glycyl-(Z)-dehydrophenyalanine (Compound 13)
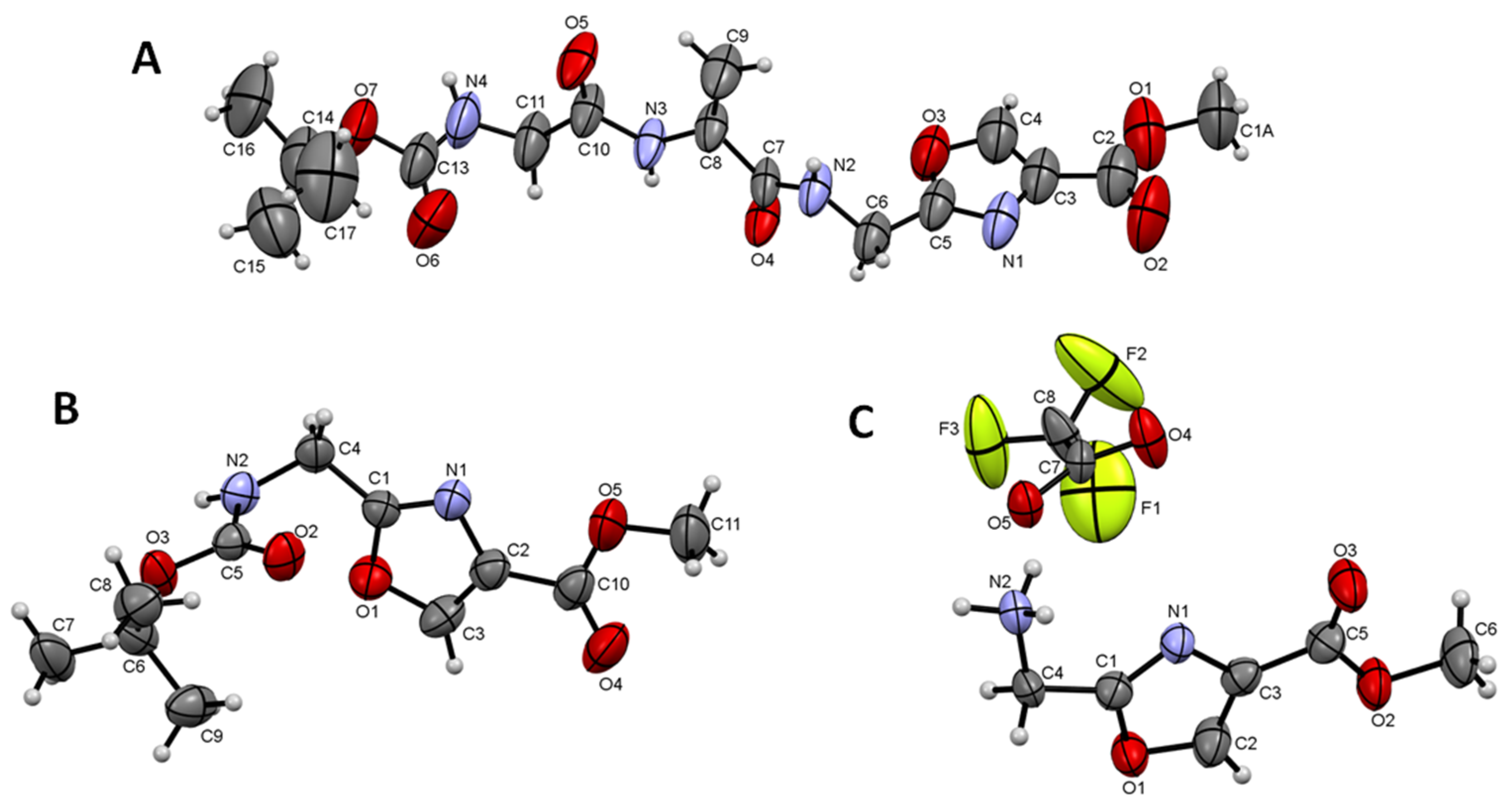
4.3. Crystallography
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Nanda, V.; Koder, R.L. Designing artificial enzymes by intuition and computation. Nat. Chem. 2010, 2, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raffy, Q.; Ricoux, R.; Mahy, J.-P. A new kind of eco-compatible hybrid biocatalyst for selective reactions: Artificial metalloenzymes. In Focus on Catalysis Research: New Developments, 1st ed.; Ghang, M., Ramel, B., Eds.; Nova Science Publishers Inc.: New York, NY, USA, 2011; pp. 31–62. [Google Scholar]
- Ward, T.R. Artificial metalloenzymes based on the biotin−avidin technology: Enantioselective catalysis and beyond. Acc. Chem. Res. 2011, 44, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Raynal, M.; Ballester, P.; Vidal-Ferran, A.; van Leeuven, P.W.N.M. Supramolecular catalysis. Part 2: Artificial enzyme mimics. Chem. Soc. Rev. 2014, 43, 1734–1787. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, T.; Ghattas, W.; Herrero, C.; Velours, C.; Minard, P.; Mahy, J.-P.; Ricoux, R.; Urvoas, A. αRep A3: A versatile artificial scaffold for metalloenzyme design. Chem. Eur. J. 2017, 23, 10156–10166. [Google Scholar] [CrossRef]
- Sanasiaume-Dagousset, E.; Urvoas, A.; Chelly, K.; Ghattas, W.; Maréchal, J.-D.; Mahy, J.P.; Ricoux, R. Neocarzinostatin-based hybrid biocatalysts for oxidation reactions. Dalton Trans. 2014, 43, 8344–8354. [Google Scholar] [CrossRef]
- Köhler, V.; Wilson, Y.M.; Lo, C.; Sardo, A.; Ward, T.R. Protein-based hybrid catalysts—design and evolution. Curr. Opin. Biotechnol. 2010, 21, 744–775. [Google Scholar] [CrossRef]
- Talbi, B.; Haquette, P.; Marel, A.; de Montigny, F.; Fosse, C.; Cordier, S.; Roisnel, T.; Jaouen, G.; Salamain, M. (η6-Arene) ruthenium (II) complexes and metallo-papain hybrid as Lewis acid catalysts of Diels–Alder reaction in water. Dalton Trans. 2010, 39, 5605–5607. [Google Scholar] [CrossRef] [Green Version]
- Bayat, S.; Tejo, B.A.; Abdulmalek, E.; Salleh, A.B.; Normi, Y.M.; Rahman, M.B.A. Rational design of mimetic peptides aldo-ketoreductase enzyme as asymmetric organocatalysts in aldol reactions. RSC Adv. 2014, 4, 38859–38868. [Google Scholar] [CrossRef]
- Yuan, Z.; Jin, Z.; Yingwu, L. Rational design of hydrolases in protein scaffolds. Prog. Chem. 2015, 27, 1102–1109. [Google Scholar]
- Razkin, J.; Nilsson, H.; Baltzer, L. Catalysis of the cleavage of uridine 3′-2,2,2-trichloroethylphosphate by a designed helix−loop−helix motif peptide. J. Am. Chem. Soc. 2007, 129, 14752–14758. [Google Scholar] [CrossRef]
- McAllister, K.A.; Zou, H.; Cochran, F.V.; Bender, G.M.; Senes, A.; Fry, H.C.; Nanda, V.; Keenan, P.A.; Lear, J.D.; Saven, J.G.; et al. Using α-helical coiled-coils to design nanostructured metalloporphyrin arrays. J. Am. Chem. Soc. 2008, 130, 11921–11927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zastrow, M.L.; Peacock, A.F.A.; Stuckey, J.A.; Pecoraro, V.L. Hydrolytic catalysis and structural stabilization in a designed metalloprotein. Nat. Chem. 2011, 4, 118–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, C.T.; Watkins, D.W.; Anderson, J.L.R. Constructing manmade enzymes for oxygen activation. Dalton Trans. 2013, 42, 3136–3150. [Google Scholar] [CrossRef] [PubMed]
- Drewniak, M.; Węglarz-Tomczak, E.; Ożga, K.; Rudzińska-Szostak, E.; Macegoniuk, K.; Tomczak, J.M.; Bejger, M.; Rypniewski, W.; Berlicki, Ł. Helix-loop-helix peptide foldamers and their use in the construction of hydrolase mimetics. Bioorg. Chem. 2018, 81, 356–361. [Google Scholar] [CrossRef]
- Ferrazzano, L.; Corbisiero, D.; Greco, R.; Potenza, E.; De Seriis, G.; Garelli, A.; Tolomelli, A. Synthesis of α/β dipeptides containing linear or cyclic α-dehydro-β-amino acids as scaffolds for bioactive compounds. Amino Acids 2019, 51, 1475–1483. [Google Scholar] [CrossRef]
- Goodman, C.M.; Choi, S.; Shandler, S.; De Grado, W.F. Foldamers as versatile frameworks for the design and evolution of function. Nat. Chem. Biol. 2007, 3, 252–262. [Google Scholar] [CrossRef] [Green Version]
- Joaquin, D.; Lee, M.A.; Kastner, D.W.; Singh, J.; Morrill, S.T.; Damestedt, G.; Castle, S.L. Impact of dehydroamino acids on the structure and stability of incipient 310-helical peptides. J. Org. Chem. 2020, 85, 1601–1613. [Google Scholar] [CrossRef]
- Jalan, A.; Kastner, D.W.; Webber, K.G.I.; Smith, M.S.; Price, J.L.; Castle, S.L. Bulky dehydroamino acids enhance proteolytic stability and folding in β-hairpin peptides. Org. Lett. 2017, 19, 5190–5193. [Google Scholar] [CrossRef]
- Shivhare, A.; Garg, C.; Priyam, A.; Gupta, A.; Sharma, A.K.; Kumar, P. Enzyme sensitive smart inulin-dehydropeptide conjugate self assembles into nanostructures useful for targeted delivery of ornidazole. Int. J. Biol. Macromol. 2018, 106, 775–783. [Google Scholar] [CrossRef]
- De Vries, R.H.; Viel, J.H.; Oudshoorn, R.; Kuipers, O.P.; Gerard Roelfes, G. Selective modification of ribosomally synthesized and post-translationally modified peptides (RiPPs) through Diels–Alder cycloadditions on dehydroalanine sesidues. Eur. J. Chem. 2019, 55, 12698–12702. [Google Scholar] [CrossRef] [Green Version]
- Broda, M.A.; Siodłak, D.; Rzeszotarska, B. Conformational investigation of α,β-dehydropeptides. XV: N-acetyl-α,β-dehydroamino acid N ′N ′-dimethylamides: Conformational properties from infrared and theoretical studies. J. Pept. Sci. 2005, 11, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Latajka, R.; Jewgiński, M.; Makowski, M.; Krężel, A. Conformational studies of hexapeptides containing two dehydroamino acid residues in positon 3 and 5 in peptide chain. J. Mol. Struct. 2008, 892, 446–451. [Google Scholar] [CrossRef]
- Buczek, A.; Makowski, M.; Jewgiński, M.; Latajka, R.; Kupka, T.; Broda, M.A. Toward engineering efficient peptidomimetics. screening conformational landscape of two modified dehydroaminoacids. Biopolymers 2014, 101, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Chao, W.; Li, L.; Zheng, R.; Sun, D. Synthesis of dehydroamino acids and their applications in the drug research and development. Progress Chem. 2020, 32, 55–71. [Google Scholar]
- Siodłak, D. α,β-Dehydroamino acids in naturally occurring peptides. Amino Acids 2015, 47, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Levengood, M.R.; van der Donk, W.A. Dehydroalanine-containing peptides: Preparation from phenylselenocysteine and utility in convergent ligation strategies. Nat. Prot. 2006, 1, 3001–3009. [Google Scholar] [CrossRef]
- Cossec, B.; Cosnier, F.; Burgart, M. Methyl mercapturate synthesis: An efficient, convenient and simple method. Molecules 2008, 13, 2394–2407. [Google Scholar] [CrossRef] [Green Version]
- Lenartowicz, P.; Makowski, M.; Oszywa, B.; Haremza, K.; Latajka, R.; Pawełczak, M.; Kafarski, P. Addition of thiols to the double bond of dipeptide C -terminal dehydroalanine as a source of new inhibitors of cathepsin C. Biochimie 2017, 139, 46–65. [Google Scholar] [CrossRef]
- Lenartowicz, P.; Dziuk, B.; Zarychta, B.; Makowski, M.; Kafarski, P. Michael additions to double bonds of esters of N-protected (s)-phenylalanyldehydroalanine (X-(s)-Phe-ΔAla-OMe) and its phosphonic acid counterpart (X-(s)-Phe-ΔAla-PO(OEt)2). Phosphorus Sulfur Silicon Relat. Elem. 2017, 192, 706–712. [Google Scholar] [CrossRef]
- Lenartowicz, P.; Psurski, M.; Kotynia, A.; Pieniężna, A.; Cuprych, M.; Poniatowska, K.; Brasuń, J.; Kafarski, P. Dipeptides of S-substituted dehydrocysteine as artzyme building blocks: Synthesis, complexing abilities and antiproliferative properties. Int. J. Mol. Sci. 2021, 22, 2168. [Google Scholar] [CrossRef]
- Bogart, J.W.; Bowers, A.A. Dehydroamino acids: Chemical multi-tools for late-stage diversification. Org. Biomol. Chem. 2019, 17, 3653–3669. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Zhu, J.; Wang, W.; Zhang, Y. Synthesis of β-silyl α-amino Acids via visible-light-mediated hydrosilylation. Org. Lett. 2021, 23, 1406–1410. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, P.M.T.; Maia, H.L.S.; Monteiro, L.S.; Sacramento, J. Michael addition of thiols, carbon nucleophiles and amines to dehydroamino acid and dehydropeptide derivatives. J. Chem. Soc. Perkin Trans. 1 2001, 3167–3173. [Google Scholar] [CrossRef]
- Ferreira, P.M.T.; Maia, H.L.S.; Monteiro, L.S. Synthesis of Non-Natural Amino Acids from N-(p-Tolylsulfonyl)-α,β-didehydroamino Acid Derivatives. Eur. J. Org. Chem. 2003, 2003, 2635–2644. [Google Scholar] [CrossRef]
- Kakkar, S.; Narasimham, B. A comprehensive review on biological activities of oxazole derivatives. BMC Chem. 2019, 13, 16. [Google Scholar] [CrossRef] [Green Version]
- Mhlongo, J.T.; Brasil, E.; de la Torre, B.G.; Albericio, F. Naturally occurring oxazole-containing peptides. Mar. Drugs 2020, 18, 203. [Google Scholar] [CrossRef]
- Petersen, J.; Christensen, K.E.; Nielsen, M.T.; Mortenses, K.T.; Komnatnyy, V.V.; Nielsen, T.E.; Qvortrup, K. Oxidative modification of tryptophan-containing peptides. ACS Comb. Sci. 2018, 20, 344–349. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Mo, J.; Lin, H.-Z.; Chen, Y.; Sun, H.-P. The recent progress of isoxazole in medicinal chemistry. Bioorg. Med. Chem. Lett. 2018, 26, 3065–3075. [Google Scholar] [CrossRef]
- Siodłak, D.; Staś, M.; Broda, M.A.; Bujak, M.; Lis, T. Conformational properties of oxazole-amino acids: Effect of the intramolecular N-H···N hydrogen bond. J. Phys. Chem. B 2014, 118, 2340–2360. [Google Scholar] [CrossRef]
- Staś, M.; Bujak, M.; Broda, M.A.; Siodłak, D. Conformational preferences and synthesis of isomers Z and E of oxazole-dehydrophenylalanine. Biopolymers 2016, 106, 283–294. [Google Scholar] [CrossRef]
- Tsutsumi, H.; Kuroda, T.; Kimura, H.; Goto, Y.; Suga, H. Posttranslational chemical installation of azoles into translated peptides. Nat. Commun. 2021, 12, 696. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, R.; Mishra, R.; Chandrasekaran, S. An improved procedure for the synthesis of dehydroamino acids and dehydropeptides from the carbonate derivatives of serine and threonine using tetrabutylammonium fluoride. J. Pep. Sci. 2010, 16, 123–126. [Google Scholar]
- Tian, X.; Li, L.; Han, J.; Zhen, X.; Liu, S. Stereoselectively synthesis and structural confirmation of dehydrodipeptides with dehydrobutyrine. SpringerPlus 2016, 5, 400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Latajka, R.; Makowski, M.; Jewgiński, M.; Pawełczak, M.; Koroniak, H.; Kafarski, P. Peptide p-nitrophenylanilides containing (E)-dehydrophenylalanine—synthesis, structural studies and evaluation of their activity towards cathepsin C. New. J. Chem. 2006, 30, 1009–1018. [Google Scholar] [CrossRef]
- Makowski, M.; Lenartowicz, P.; Oszywa, B.; Jewgiński, M.; Pawełczak, M.; Kafarski, P. Synthesis of dehydrodipeptide esters and their evaluation as inhibitors of cathepsin C. Med. Chem. Res. 2015, 24, 3157–3165. [Google Scholar] [CrossRef] [Green Version]
- Shin, C.G.; Yonezawa, Y.; Takahashi, M.; Yoshimura, J. Dehydropeptides II. The synthesis of dehydrodipeptides by direct coupling and base-catalyzed β-elimination. Bull. Soc. Chem. Jpn. 1981, 54, 1132–1136. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, P.M.T.; Maia, G.L.S.; Monteiro, L.S.; Sacramento, J. High yielding synthesis of dehydroamino acid and dehydropeptide derivatives. J. Chem. Soc. Perkin Trans. 1 1999, 24, 3697–3703. [Google Scholar] [CrossRef]
- Ramesh, R.; De, K.; Chandrasekaran, S. An efficient synthesis of dehydroamino acids and dehydropeptides from O-Cbz and O-Fmoc derivatives of serine and threonine. Tetrahedron 2007, 63, 10534–10542. [Google Scholar] [CrossRef]
- Makowski, M.; Rzeszotarska, B.; Kubica, Z.; Pietrzyński, G. Synthesis of Peptides with α,β-Dehydroamino Acids, II. Synthesis of tert-butyloxycarbonyldipeptides of dehydroalanine and dehydrophenylalanine. Liebigs Ann. Chem. 1985, 5, 893–900. [Google Scholar] [CrossRef]
- Makowski, M.; Rzeszotarska, B.; Kubica, Z.; Wieczorek, P.; Makowski, M.; Rzeszotarska, B.; Kubica, Z.; Wieczorek, P. Synthesis of Peptides with α,β-Dehydroamino Acids, I Synthesis of N-benzyloxycarbonyl and N-trifluoroacetyl dipeptides of dehydroalanine and dehydrophenylalanine. Liebigs Ann. Chem. 1984, 5, 920–928. [Google Scholar] [CrossRef]
- Murru, S.; Nefzi, A. Combinatorial synthesis of oxazol-thiazole bis-heterocyclic compounds. ACS Comb. Sci. 2014, 16, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.J.; Uto, Y.; Wipf, P.; Reno, M.J.; Williams, D.R. Synthesis of functionalized oxazolines and oxazoles with DAST and Deoxo-Fluor. Org. Lett. 2000, 2, 1165–1168. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, D.; Videnov, G.; Maichle-Mossmer, C.; Strahle, J.; Jung, G. X-Ray structures and ab initio study of the conformational properties of novel oxazole and thiazole containing di- and tripeptide mimetics. J. Chem. Soc. Perkin Trans. 2 2000, 5, 1081–1085. [Google Scholar] [CrossRef]
- Latajka, R.; Jewgiński, M.; Makowski, M.; Krężel, A. Conformational studies of hexapeptides containing two dehydroamino acid residues in positon 2 and 5 in peptide chain. Biopolymers 2008, 89, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Gruß, H.; Feiner, R.C.; Mseya, R.; Schröder, D.C.; Jewgiński, M.; Müller, K.M.; Latajka, R.; Marion, A.; Seewald, N. Peptide stapling by late-stage Suzuki–Miyaura cross-coupling. Beilstein J. Org. Chem. 2022, 18, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Jewgiński, M.; Latajka, R.; Krężel, A.; Haremza, K.; Makowski, M.; Kafarski, P. Influence of solvents on conformation of dehydropeptides. J. Mol. Struct. 2013, 1035, 129–139. [Google Scholar] [CrossRef]
- Jewgiński, M.; Krzciuk-Gula, J.; Makowski, M.; Latajka, R.; Kafarski, P. Conformation of dehydropentapeptides containing four achiral amino acid residues—controlling the role of L-valine. Beilstein J. Org. Chem. 2014, 10, 660–666. [Google Scholar] [CrossRef] [Green Version]
- Jaremko, M.; Jaremko, Ł.; Mazur, A.; Makowski, M.; Lisowski, M.; Mazur, A. Enhanced β-turn conformational stability of tripeptides containing ΔPhe in cis over trans configuration. Amino Acids 2013, 45, 865–875. [Google Scholar] [CrossRef]
- CrysAlis CCD, Version 1.171.33.57; Oxford Diffraction Ltd.: Abingdon, UK, 2005.
- CrysAlis RED, Version 1.171.33.57; Oxford Diffraction Ltd.: Abingdon, UK, 2005.
- Rigaku Oxford Difraction. CrysAlisPro; Rigaku Oxford Diffraction: Wroclaw, Poland, 2016. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. New features added to the refinement program SHELXL since 2008 are described and explained. Acta Crystallogr. Sect. C 2015, 71, 13–18. [Google Scholar]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. New Features for the Visualization and Investigation of Crystal Structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Synthetic Method | Isolated Yield | |
|---|---|---|---|
| 1a | Dehydration of serine residues (MsCl, Et3N, Et3N × HCl, DBU in DMF at −10 °C, 2.5 h) | 70% 1 | |
| 1b | 73% | ||
| 1c | Coupling of dipeptide building blocks—mixed anhydride method (Isobutyl chloroformate, NMM in ACN or THF at −15 °C, overnight) | 48% | |
| 3 | 55% | ||
| 4 | Coupling of dipeptide building blocks—guanidinium salt method (TBTU, Et3N in ACN at −10 °C, 12 h) | 66% | |
| 6a | 59% 2 | ||
| 6b | 56% 2 | ||
| 10 | Acylation of dipeptide building block by acyl fluoride (1. TFFH, DIPEA in DCM at 8–9 °C, 0.5 h; 2. Boc-AA-ΔAA-F, Et3N in DCM at rt, 4 h) | 21% | Total 74% 3 |
| 11 | 53% | ||
| 12 | 24% | Total 71% 3 | |
| 13 | 47% | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lenartowicz, P.; Beelen, M.; Makowski, M.; Wanat, W.; Dziuk, B.; Kafarski, P. Synthesis of Tetrapeptides Containing Dehydroalanine, Dehydrophenylalanine and Oxazole as Building Blocks for Construction of Foldamers and Bioinspired Catalysts. Molecules 2022, 27, 2611. https://doi.org/10.3390/molecules27092611
Lenartowicz P, Beelen M, Makowski M, Wanat W, Dziuk B, Kafarski P. Synthesis of Tetrapeptides Containing Dehydroalanine, Dehydrophenylalanine and Oxazole as Building Blocks for Construction of Foldamers and Bioinspired Catalysts. Molecules. 2022; 27(9):2611. https://doi.org/10.3390/molecules27092611
Chicago/Turabian StyleLenartowicz, Paweł, Maarten Beelen, Maciej Makowski, Weronika Wanat, Błażej Dziuk, and Paweł Kafarski. 2022. "Synthesis of Tetrapeptides Containing Dehydroalanine, Dehydrophenylalanine and Oxazole as Building Blocks for Construction of Foldamers and Bioinspired Catalysts" Molecules 27, no. 9: 2611. https://doi.org/10.3390/molecules27092611
APA StyleLenartowicz, P., Beelen, M., Makowski, M., Wanat, W., Dziuk, B., & Kafarski, P. (2022). Synthesis of Tetrapeptides Containing Dehydroalanine, Dehydrophenylalanine and Oxazole as Building Blocks for Construction of Foldamers and Bioinspired Catalysts. Molecules, 27(9), 2611. https://doi.org/10.3390/molecules27092611