
Synthesis, Experimental and Theoretical Study of Azidochromones

 , , , and
, , , and
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Conformational Analysis
2.2. NMR Spectroscopy
2.3. Electronic Spectra
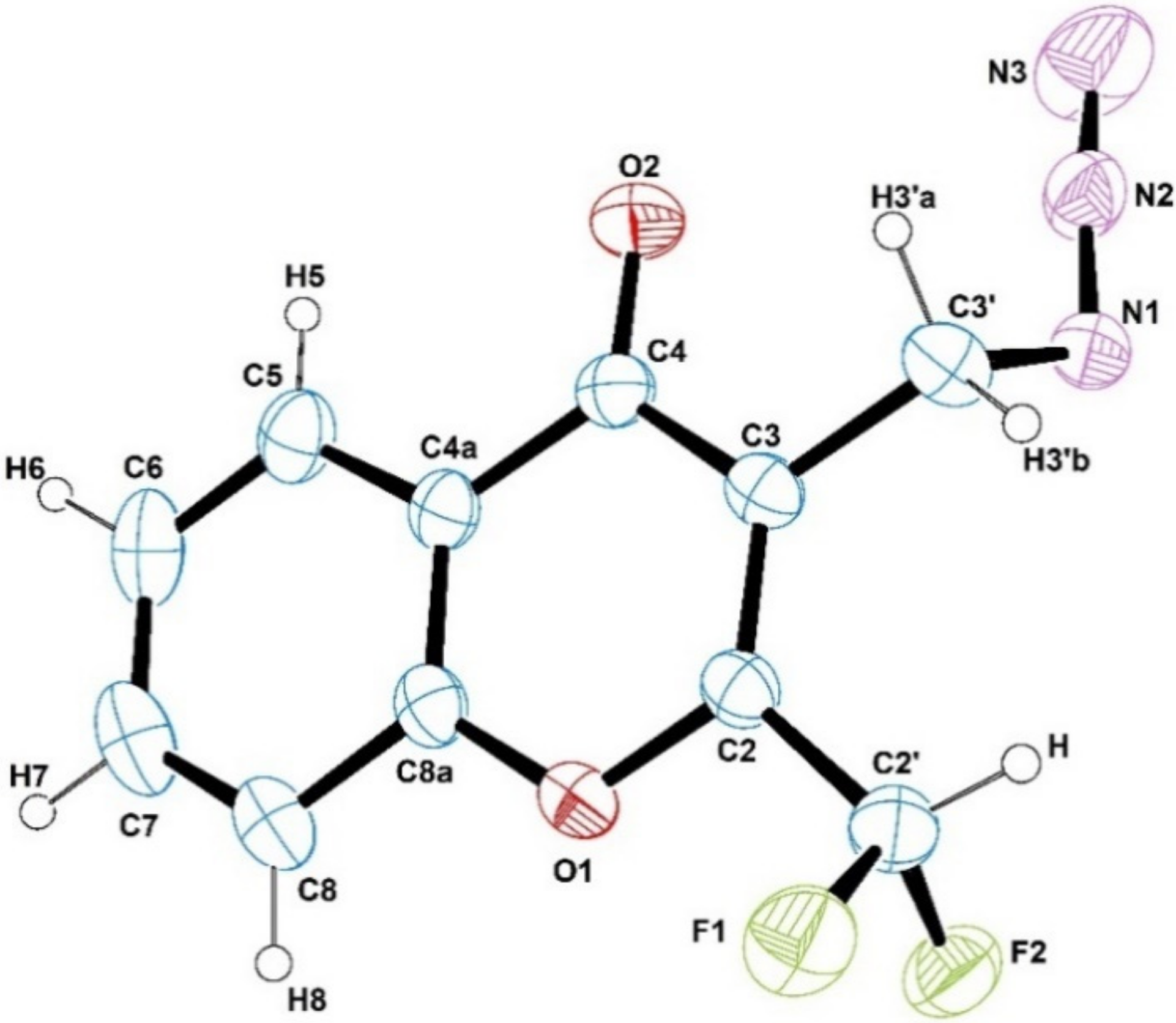
2.4. Crystallographic Structural Results of (4) or: Crystal Structure of 4
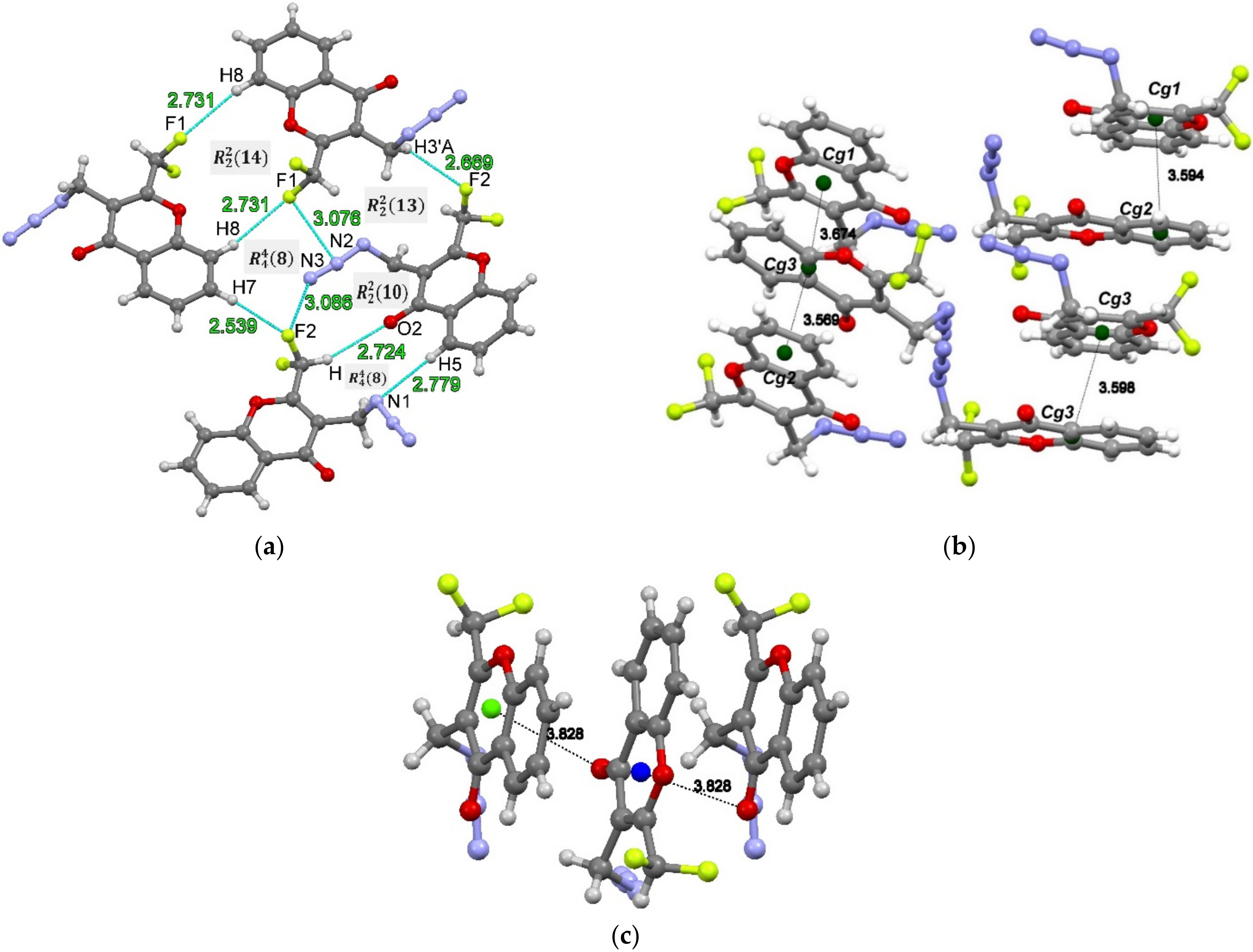
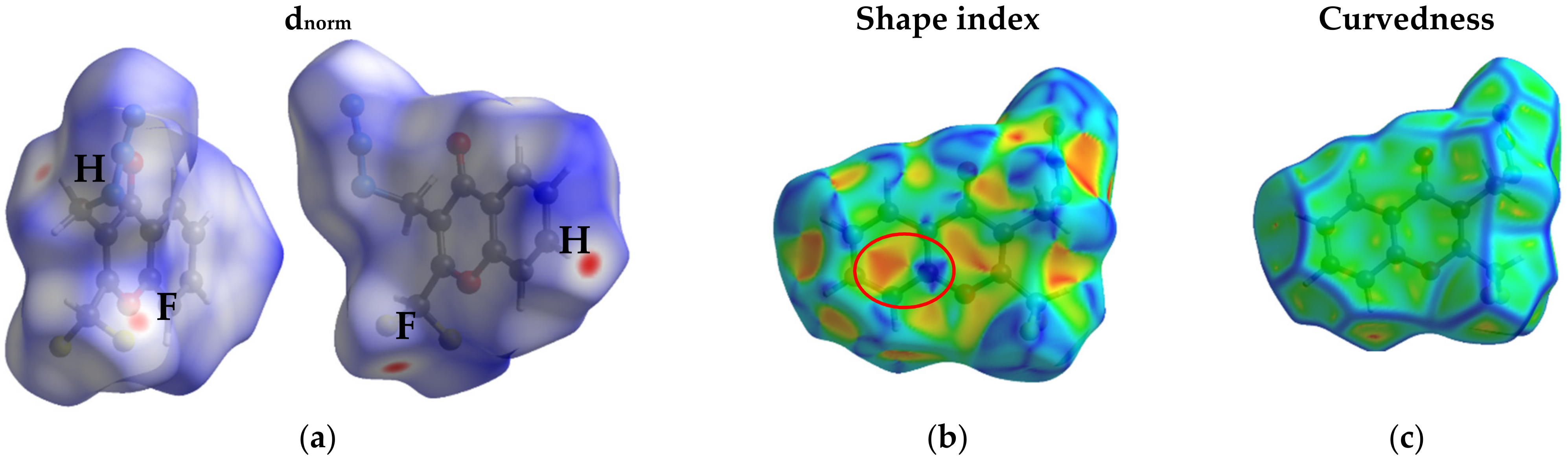
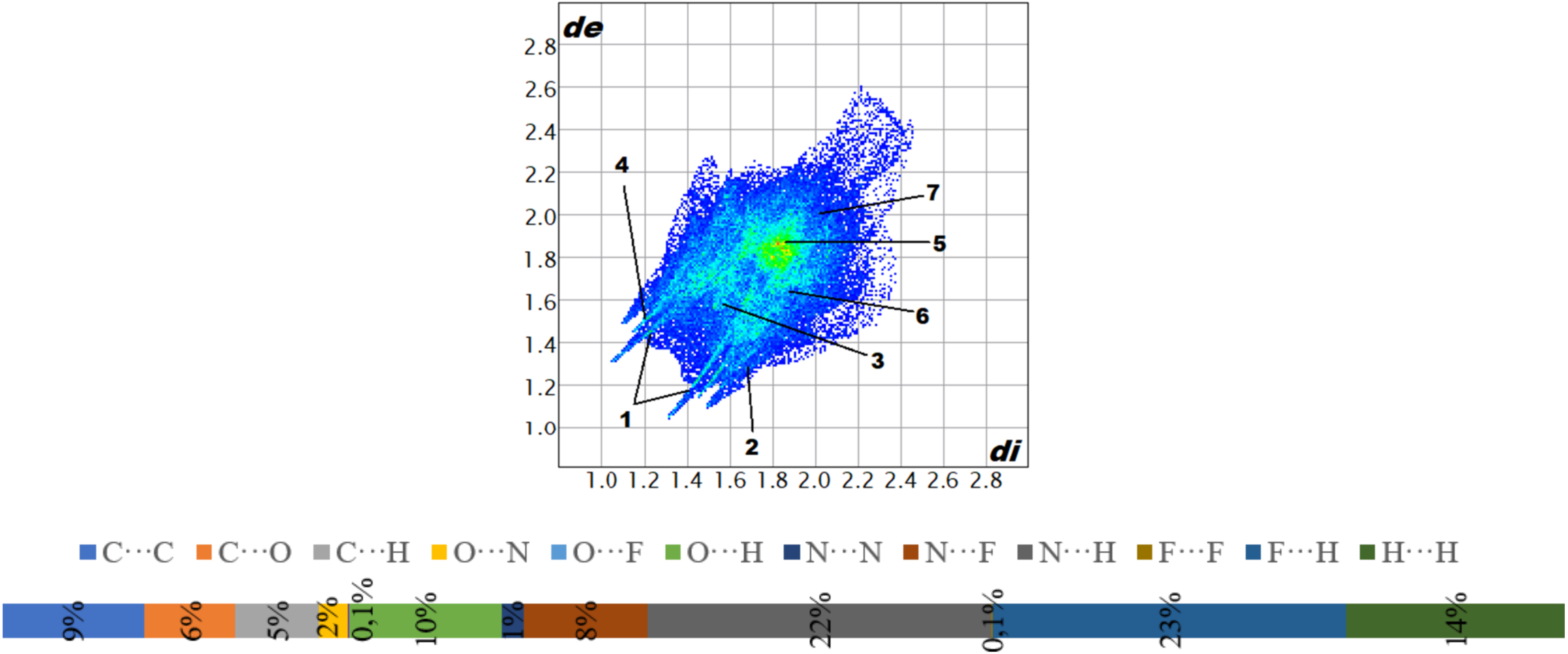
2.5. Hirshfeld Surface Analysis
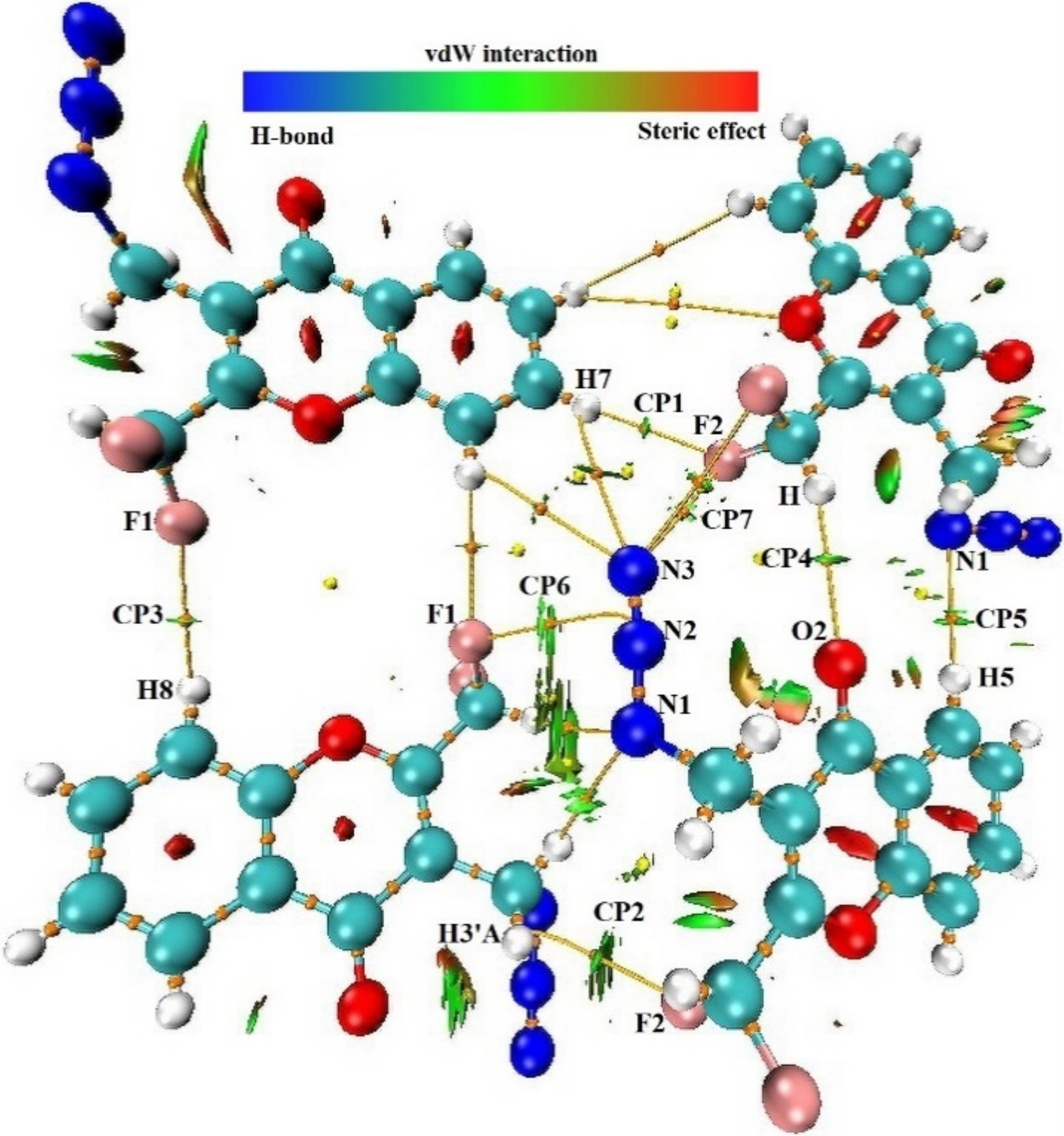
2.6. AIM and NCI Analysis of Intermolecular Contacts of 4
3. Experimental
3.1. General
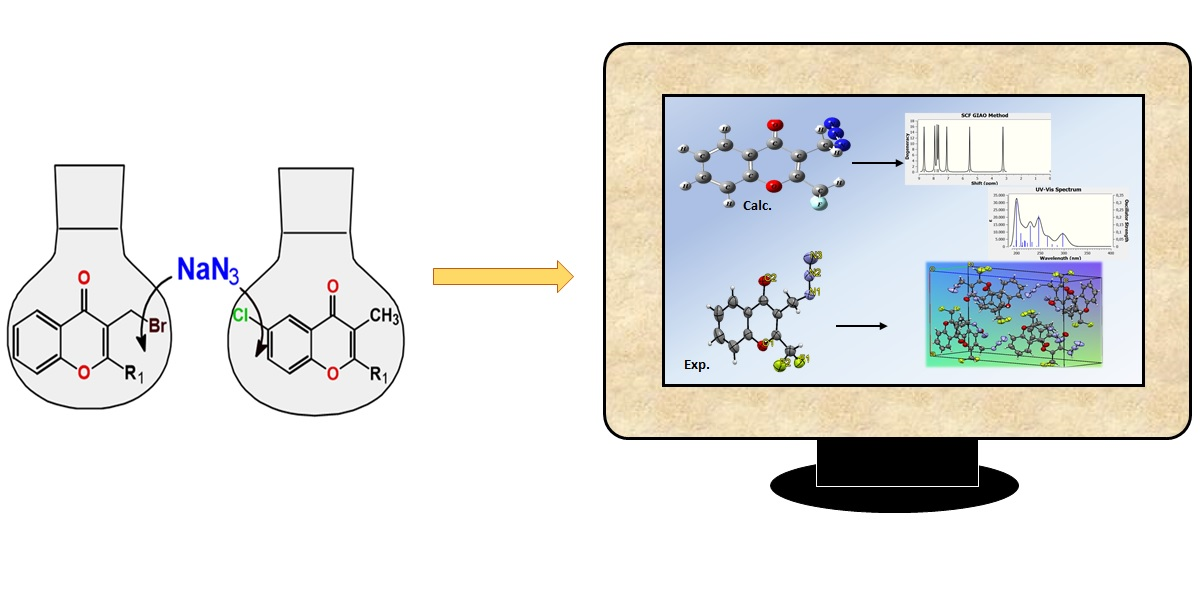
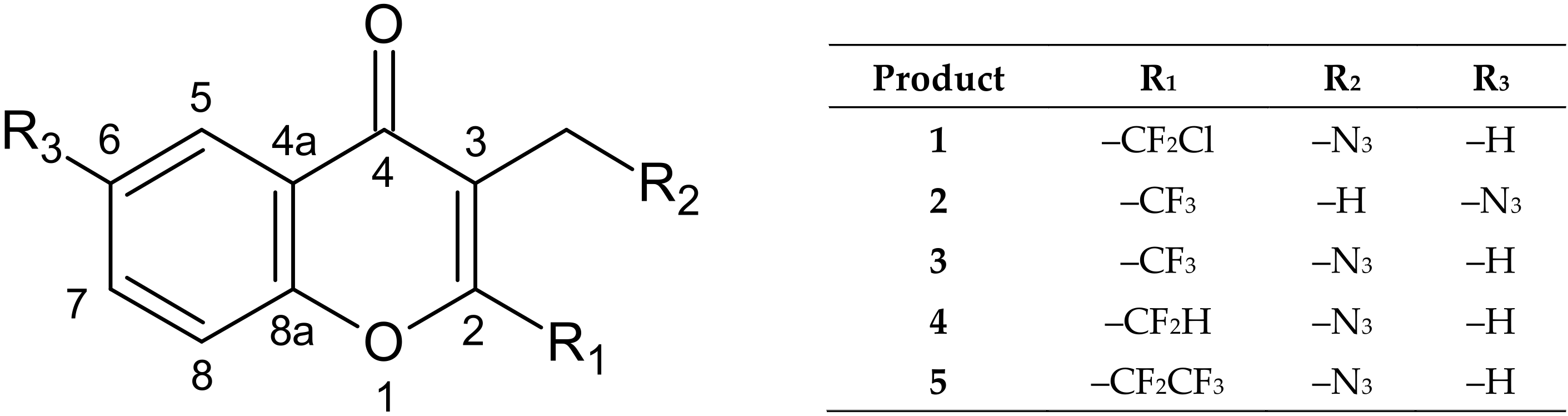
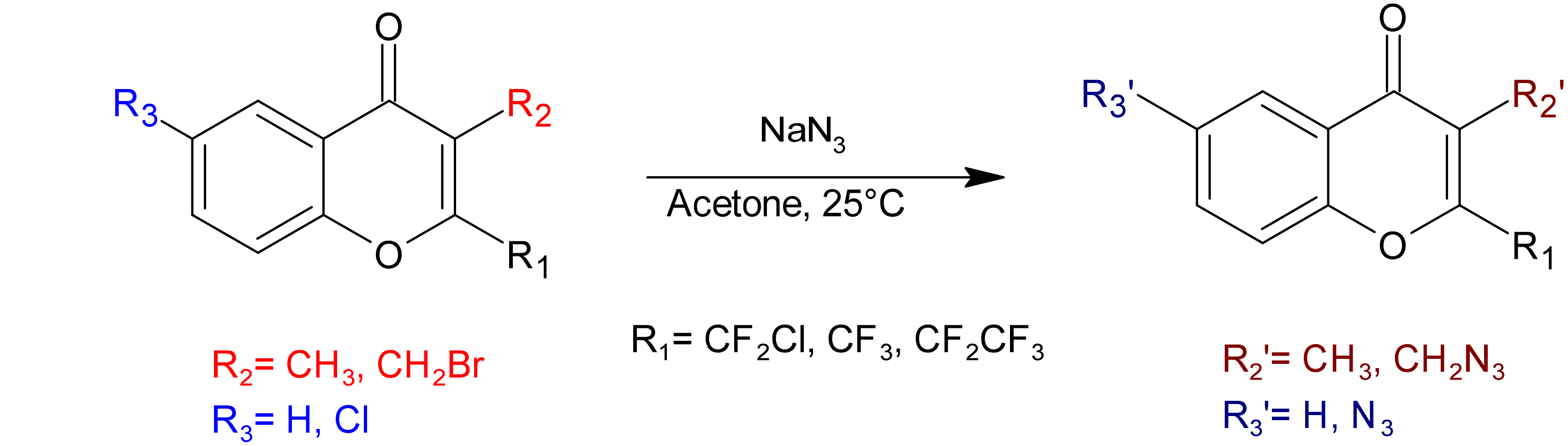
3.2. Synthesis: General Procedure for Azidochromones (1–5)
3.3. X ray Diffraction Date and Structural Refinement of 4
3.4. Computational Details
3.5. Hirshfeld Surface Calculations of 4
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Sosnovskikh, V.Y. Synthesis and Reactions of Halogen-Containing Chromones. Russ. Chem. Rev. 2003, 72, 489–516. [Google Scholar] [CrossRef]
- Sosnovskikh, V.Y.; Korotaev, V.Y.; Chizhov, D.L.; Kutyashev, I.B.; Yachevskii, D.S.; Kazheva, O.N.; Dyachenko, O.A.; Charushin, V.N. Reaction of Polyhaloalkyl-Substituted Chromones, Pyrones, and Furanones with Salicylaldehydes as a Direct Route to Fused 2H-Chromenes. J. Org. Chem. 2006, 71, 4538–4543. [Google Scholar] [CrossRef] [PubMed]
- Alcívar León, C.D.; Echeverría, G.A.; Piro, O.E.; Ulic, S.E.; Jios, J.L.; Pereañez, J.A.; Henao Castañeda, I.C.; Pérez, H. The Role of Non-Covalent Interactions in Some 2-Trifluoromethylchromones in the Solid State. New J. Chem. 2017, 41, 14659–14674. [Google Scholar] [CrossRef]
- Alcívar León, C.D. Estudio de Nuevos Benzopiranos Haloalquil Sustituidos. PhD Thesis, Universidad Nacional de La Plata, La Plata, Argentina, 2016. [Google Scholar]
- Narváez, O.E.G.; Bonilla, V.P.M.; Zurita, D.A.; Alcívar, L.C.D.; Heredia-Moya, J.; Ulic, S.E.; Jios, J.L.; Piro, O.E.; Echeverría, G.A.; Langer, P. Synthesis, Experimental and Theoretical Study of Novel 2-Haloalkyl (-CF2H, -CCl2H, -CF2CF3)-, 3-Bromo and Bromomethyl Substituted Chromones. J. Fluor. Chem. 2021, 242, 109717. [Google Scholar] [CrossRef]
- Gomes, A.; Neuwirth, O.; Freitas, M.; Couto, D.; Ribeiro, D.; Figueiredo, A.G.P.R.; Silva, A.M.S.; Seixas, R.S.G.R.; Pinto, D.C.G.A.; Tomé, A.C.; et al. Synthesis and Antioxidant Properties of New Chromone Derivatives. Bioorg. Med. Chem. 2009, 17, 7218–7226. [Google Scholar] [CrossRef]
- Fitton, A.O.; Hatton, B.T. Pharmacologically Active 4-Oxo-4H-Chromen-2-Carboxylic Acids. Part II. The Synthesis of 4-Oxo-4H-Chromen-2-Carboxylic Acids Containing a Fused Imidazole or Oxazole Ring. J. Chem. Soc. C Org. 1970, 2518–2522. [Google Scholar] [CrossRef]
- Chohan, M.I.; Fitton, A.O.; Hatton, B.T.; Suschitzky, H. Pharmacologically Active 4-Oxo-4H-Chromen-2-Carboxylic Acids. Part III. Mechanistic Aspects of the Decomposition of Ethyl 6-Azido-4-Oxo-4H-Chromen-2-Carboxylate. J. Chem. Soc. C Org. 1971, 3079–3081. [Google Scholar] [CrossRef]
- Tome, S.M.; Tome, A.C.; Soengas, R.G.; Silva, A.M.S. Chalcones and Chromones in Copper-Catalyzed Azide–Alkyne Cycloadditions (CuAAC). Curr. Org. Chem. 2018, 22, 1307–1325. [Google Scholar] [CrossRef]
- Machado, N.F.L.; Marques, M.P.M. Bioactive Chromone Derivatives—Structural Diversity. Curr. Bioact. Compd. 2010, 6, 76–89. [Google Scholar] [CrossRef] [Green Version]
- Ellis, G.P. Chromones. In Chemistry of Heterocyclic Compounds: Chromenes, Chromanones, and Chromones; John Wiley & Sons, Inc.: Toronto, ON, Canada, 1977; Volume 31, p. 613. ISBN 0-471-38212-4. [Google Scholar]
- Sharma, S.; Kumar, S.; Chand, K.; Kathuria, A.; Gupta, A.; Jain, R. An Update on Natural Occurrence and Biological Activity of Chromones. Curr. Med. Chem. 2011, 18, 3825–3852. [Google Scholar] [CrossRef]
- Khadem, S.; Marles, R.J. Chromone and Flavonoid Alkaloids: Occurrence and Bioactivity. Mol. Basel Switz. 2011, 17, 191–206. [Google Scholar] [CrossRef] [PubMed]
- Tome, S.M.; Silva, A.M.S.; Santos, C.M.M. Synthesis and Transformation of Halochromones. Curr. Org. Synth. 2014, 11, 317–341. [Google Scholar] [CrossRef] [Green Version]
- Bräse, S.; Banert, K. Lab—Scale Synthesis of Azido Compounds: Safety Measures and Analysis. In Organic Azides: Syntheses and Applications; Wiley: Chichester, UK, 2010; pp. 3–4. ISBN 9780470519981. [Google Scholar]
- Wen, L.; Zhang, H.; Lin, H.; Shen, Q.; Lu, L. A Facile Synthetic Route to 2-Trifluoromethyl-Substituted Polyfunctionalized Chromenes and Chromones. J. Fluor. Chem. 2012, 133, 171–177. [Google Scholar] [CrossRef]
- Sosnovskikh, V.Y.; Irgashev, R.A.; Usachev, B.I. Synthesis and Some Properties of 2-(Polyfluoroalkyl)Chroman-4-Ols and 2-(Polyfluoroalkyl)Chroman-4-Ones. Russ. Chem. Bull. 2009, 58, 2465–2473. [Google Scholar] [CrossRef]
- Chang, M.-Y.; Tsai, Y.-L. Stereocontrolled Synthesis of 3-Sulfonyl Chroman-4-Ols. J. Org. Chem. 2018, 83, 6798–6804. [Google Scholar] [CrossRef]
- Malets, Y.S.; Moskvina, V.S.; Grygorenko, O.O.; Brovarets, V.S. Synthesis of Azachromones and Azachromanones. Chem. Heterocycl. Compd. 2019, 55, 1007–1012. [Google Scholar] [CrossRef]
- Duan, Y.; Jiang, Y.; Guo, F.; Chen, L.; Xu, L.; Zhang, W.; Liu, B. The Antitumor Activity of Naturally Occurring Chromones: A Review. Fitoterapia 2019, 135, 114–129. [Google Scholar] [CrossRef]
- Patil, V.M.; Masand, N.; Verma, S.; Masand, V. Chromones: Privileged Scaffold in Anticancer Drug Discovery. Chem. Biol. Drug Des. 2021, 98, 943–953. [Google Scholar] [CrossRef]
- Benny, A.T.; Arikkatt, S.D.; Vazhappilly, C.G.; Kannadasan, S.; Thomas, R.; Leelabaiamma, M.S.N.; Radhakrishnan, E.K.; Shanmugam, P. Chromone a Privileged Scaffold in Drug Discovery: Developments on the Synthesis and Bioactivity. Mini Rev. Med. Chem. 2021. [CrossRef]
- Sosnovskikh, V.Y. Fluorinated Pyrones, Chromones and Coumarins BT—Fluorine in Heterocyclic Chemistry. In 6-Membered Heterocycles; Nenajdenko, V., Ed.; Springer International Publishing: Cham, Switzerland, 2014; Volume 2, pp. 211–290. ISBN 978-3-319-04435-4. [Google Scholar]
- Alcívar León, C.D.; Echeverría, G.A.; Piro, O.E.; Ulic, S.E.; Jios, J.L.; Burgos Paci, M.; Argüello, G.A. The Role of Halogen C–X1⋯X2–C Contact on the Preferred Conformation of 2-Perhalomethylchromones in Solid State. Chem. Phys. 2016, 472, 142–155. [Google Scholar] [CrossRef]
- Alcívar León, C.D.; Ramos Guerrero, L.A.; Bonilla Valladares, P.M.; Echeverría, G.A.; Piro, O.E.; Ulic, S.E.; Jios, J.L.; Langer, P. Synthesis and Structural Study of 2-(Haloalkyl)-3-Methylchromones. Mon. Für Chem. Chem. Mon. 2019, 150, 1929–1940. [Google Scholar] [CrossRef]
- Alcívar León, C.D.; Piro, O.E.; Echeverria, G.A.; Ulic, S.E.; Jios, J.L. Synthesis and Conformational Theoretical Study of Novel Derivatives of 2-(Trifluoromethyl)Chromone: 3-Cyanomethyl and 3-Aminomethyl 2-(Trifluoromethyl)Chromones. J. Argent. Chem. Soc. 2014, 101, 1207–1852. [Google Scholar]
- Alcívar León, C.D.; Echeverría, G.A.; Piro, O.E.; Ulic, S.E.; Jios, J.L. A Detailed Experimental and Theoretical Study of Two Novel Substituted Trifluoromethylchromones. The Influence of the Bulky Bromine Atom on the Crystal Packing. Spectrochim. Acta Part Mol. Biomol. Spectrosc. 2015, 136, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, L. ORTEP-3 for Windows—A Version of ORTEP-III with a Graphical User Interface (GUI). J. Appl. Crystallogr. 1997, 30, 565. [Google Scholar] [CrossRef]
- Batsanov, A. Weak Interactions in Crystals: Old Concepts, New Developments. Acta Crystallogr. Sect. E Crystallogr. Commun. 2018, 74, 570–574. [Google Scholar] [CrossRef] [Green Version]
- Seth, S.K.; Sarkar, D.; Kar, T. Use of π–π Forces to Steer the Assembly of Chromone Derivatives into Hydrogen Bonded Supramolecular Layers: Crystal Structures and Hirshfeld Surface Analyses. CrystEngComm 2011, 13, 4528–4535. [Google Scholar] [CrossRef]
- Li, P.; Vik, E.C.; Maier, J.M.; Karki, I.; Strickland, S.M.S.; Umana, J.M.; Smith, M.D.; Pellechia, P.J.; Shimizu, K.D. Electrostatically Driven CO−π Aromatic Interactions. J. Am. Chem. Soc. 2019, 141, 12513–12517. [Google Scholar] [CrossRef]
- Spackman, P.; Turner, M.; McKinnon, J.; Wolff, S.; Grimwood, D.; Jayatilaka, D.; Spackman, M. CrystalExplorer: A Program for Hirshfeld Surface Analysis, Visualization and Quantitative Analysis of Molecular Crystals. J. Appl. Crystallogr. 2021, 54, 1006–1011. [Google Scholar] [CrossRef]
- Spackman, M.A.; Jayatilaka, D. Hirshfeld Surface Analysis. CrystEngComm 2009, 11, 19–32. [Google Scholar] [CrossRef]
- Bader, R.F.W. A Quantum Theory of Molecular Structure and Its Applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Boto, R.A.; Contreras-García, J.; Tierny, J.; Piquemal, J.-P. Interpretation of the Reduced Density Gradient. Mol. Phys. 2016, 114, 1406–1414. [Google Scholar] [CrossRef] [Green Version]
- Bursch, M.; Kunze, L.; Vibhute, A.M.; Hansen, A.; Sureshan, K.M.; Jones, P.G.; Grimme, S.; Werz, D.B. Quantification of Noncovalent Interactions in Azide–Pnictogen, –Chalcogen, and –Halogen Contacts. Chem. Eur. J. 2021, 27, 4627–4639. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Chen, F. Multiwfn: A Multifunctional Wavefunction Analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef] [PubMed]
- Koch, U.; Popelier, P.L.A. Characterization of C-H-O Hydrogen Bonds on the Basis of the Charge Density. J. Phys. Chem. 1995, 99, 9747–9754. [Google Scholar] [CrossRef]
- Rozas, I.; Alkorta, I.; Elguero, J. Behavior of Ylides Containing N, O, and C Atoms as Hydrogen Bond Acceptors. J. Am. Chem. Soc. 2000, 122, 11154–11161. [Google Scholar] [CrossRef]
- Espinosa, E.; Alkorta, I.; Elguero, J.; Molins, E. From Weak to Strong Interactions: A Comprehensive Analysis of the Topological and Energetic Properties of the Electron Density Distribution Involving X–H⋯F–Y Systems. J. Chem. Phys. 2002, 117, 5529–5542. [Google Scholar] [CrossRef]
- CrysAlisPRO Including ABSPACK 2009; Oxford Diffraction /Agilent Technologies UK Ltd.: Yarnton, UK, 2009.
- Sheldrick, G. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G. A Short History of SHELX. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision A.02 2009; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Mackenzie, C.F.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer Model Energies and Energy Frameworks: Extension to Metal Coordination Compounds, Organic Salts, Solvates and Open-Shell Systems. IUCrJ 2017, 4, 575–587. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Conformation | (C2-C3-C3’-X) a,c | (C3-C2-C2’-Y) b,c | % Population |
|---|---|---|---|---|
| 1 | 1 a | 95 (ac) c | 94 (ac) | 51 |
| 1 b | −81 (−sc) | 106 (ac) | 26 | |
| 1 c | −102 (−ac) | 82 (sc) | 13 | |
| 1 d | −66 (−sc) | 104 (ac) | 10 | |
| 2 | 2 | 102 (ac) | −55 (−sc) | 100 |
| 3 | 3 a | 88 (sc) | −8 (sp) | 66 |
| 3 b | 101 (ac) | 31 (sc) | 34 | |
| 4 | 4 a | 64 (sc) | 113 (ac) | 84 |
| 4 b | −105 (−ac) | −62 (−sc) | 16 | |
| 5 | 5 a | 98 (ac) | 90 (sc) | 67 |
| 5 b | 107 (ac) | −87 (−sc) | 17 | |
| 5 c | 102 (ac) | −175 (−ap) | 16 |
| 1 | 2 | 3 | 4 | 5 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Atom | Exp. | Calc. | (Δ) | Exp. | Calc. | Δ | Exp. | Calc. | Δ | Exp. | Calc. | Δ | Exp. | Calc. | Δ |
| CH3 | --- | --- | --- | 2.25 | 1.98 | 0.27 | --- | --- | --- | --- | --- | --- | --- | --- | --- |
| CH2N3 | 4.53 | 5.27 | −0.74 | --- | --- | --- | 4.46 | 4.51 | −0.05 | 4.50 | 4.44 | 0.06 | 4.46 | 4.51 | −0.05 |
| CHF2 | --- | --- | --- | --- | --- | --- | --- | --- | --- | 7.09 | 7.10 | −0.01 | --- | --- | --- |
| H-5 | 8.27 | 8.59 | −0.32 | 8.19 | 7.98 | 0.21 | 8.25 | 8.66 | −0.41 | 8.18 | 8.67 | −0.49 | 8.26 | 8.67 | −0.41 |
| H-6 | 7.53 | 7.56 | −0.03 | --- | --- | --- | 7.50 | 7.70 | −0.2 | 7.55 | 7.67 | −0.12 | 7.56 | 7.70 | −0.14 |
| H-7 | 7.82 | 7.85 | −0.03 | 7.69 | 7.58 | 0.11 | 7.79 | 7.94 | −0.15 | 7.87 | 7.97 | −0.10 | 7.80 | 7.99 | −0.19 |
| H-8 | 7.59 | 7.57 | 0.02 | 7.49 | 7.39 | 0.10 | 7.55 | 7.71 | −0.16 | 7.66 | 7.82 | −0.16 | 7.50 | 7.70 | −0.20 |
| C-2 | 154.5 | 161.0 | −6.5 | 148.5 | 155.0 | −6.5 | 151.0 | 157.7 | −6.7 | 157.2 | 163.8 | −6.6 | 150.5 | 156.5 | −6.0 |
| C-3 | 118.4 | 127.0 | −8.6 | 134.9 | 124.0 | 10.9 | 119.0 | 127.8 | −8.8 | 119.8 | 125.6 | −5.8 | 121.5 | 132.5 | −11.0 |
| C-4 | 177.1 | 188.0 | −10.9 | 176.8 | 186.0 | −9.2 | 176.9 | 182.0 | −5.1 | 178.7 | 180.2 | −1.5 | 176.8 | 182.0 | −5.2 |
| C-4a | 122.6 | 128.0 | −5.4 | 131.9 | 128.0 | 3. | 122.9 | 128.6 | −5.7 | 124.0 | 129.5 | −5.5 | 122.7 | 128.9 | −6.2 |
| C-5 | 126.6 | 131.0 | −4.4 | 119.9 | 133.0 | −13.1 | 126.4 | 132.7 | −6.3 | 127.5 | 132.5 | −5.0 | 126.9 | 132.5 | −5.6 |
| C-6 | 126.3 | 126.0 | 0.3 | 136.0 | 129.0 | 7.0 | 126.8 | 130.7 | −3.9 | 126.7 | 131.4 | −4.7 | 126.5 | 130.4 | −3.9 |
| C-7 | 135.3 | 140.0 | −4.7 | 125.4 | 128.0 | −2.6 | 135.5 | 139.6 | −4.1 | 136.6 | 139.7 | −3.1 | 135.5 | 139.1 | −3.6 |
| C-8 | 116.8 | 121.0 | −4.2 | 123.2 | 123.0 | 0.2 | 118.5 | 122.1 | −3.6 | 119.6 | 122.6 | −3.0 | 118.4 | 121.6 | −3.2 |
| C-8a | 154.9 | 164.0 | −9.1 | 153.3 | 161.0 | −7.7 | 155.2 | 162.0 | −6.8 | 157.0 | 162.5 | −5.5 | 155.4 | 162.2 | −6.8 |
| CH3 | --- | --- | --- | 8.7 | 11.4 | −2.7 | --- | --- | --- | --- | --- | --- | --- | --- | --- |
| CH2N3 | 42.8 | 45.5 | −2.70 | --- | --- | --- | 42.8 | 45.6 | −2.8 | 43.3 | 44.4 | −1.1 | 42.9 | 48.4 | −5.5 |
| CF2Cl | 120.8 | 153.0 | −32.2 | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- |
| CF3 | --- | --- | --- | 120.9 | 143.0 | −22.1 | 119.4 | 131.4 | −12.0 | --- | --- | --- | --- | --- | --- |
| CF2H | --- | --- | --- | --- | --- | --- | --- | --- | --- | 111.0 | 117.0 | −6.0 | --- | --- | --- |
| CF2CF3 | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | 120.0 | --- |
| CF2CF3 | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | --- | 129.9 | --- |
| Compound | Experimental a | Calculated b | Assignment |
|---|---|---|---|
| 1 | 204 (4.62) | 211 (0.177) | HOMO − 1 → LUMO + 3 (51%) |
| 225 (4.42) | 237 (0.136) | HOMO − 5 → LUMO (49%) | |
| 244 (4.23) | 289 (0.121) | HOMO − 2 → LUMO (88%) | |
| 303 (4.00) | 330 (0.076) | HOMO → LUMO (74%) | |
| 2 | 203 (3.87) | 212 (0.228) | HOMO → LUMO + 5 (25%) |
| 230 (3.81) | 271 (0.599) | HOMO − 2 → LUMO (41%) | |
| 318 (3.24) | 361 (0.116) | HOMO → LUMO (88%) | |
| 3 | 204 (4.55) | 201 (0.185) | HOMO – 2 →LUMO + 2 (43%) |
| 223 c (4.34) | 223 (0.103) | HOMO – 5 → LUMO + 1 (50%) | |
| 250 c (4.06) | 250 (0.230) | HOMO → LUMO + 1(37%) | |
| 305 (3.95) | 304 (0.091) | HOMO → LUMO (95%) | |
| 4 | 204 (4.63) | 204 (0.306) | HOMO – 2 → LUMO + 2 (44%) |
| 224 (4.56) | 233 (0.140) | HOMO → LUMO + 2 (37%) | |
| HOMO – 4 → LUMO (32%) | |||
| 246 c (4.36) | 250 (0.210) | HOMO → LUMO + 1 (64%) | |
| 301 (4.14) | 300 (0.093) | HOMO → LUMO (80%) | |
| 5 | 204 (4.65) | 203 (0.202) | HOMO – 2 → LUMO + 2 (50%) |
| 221 (4.46) | 223 (0.079) | HOMO – 5 → LUMO (59%) | |
| 246 (4.35) | 238 (0.108) | HOMO – 4 → LUMO (72%) | |
| 249 (0.233) | HOMO → LUMO + 1 (65%) | ||
| 271 (0.084) | HOMO – 2 → LUMO (75%) | ||
| 304 (4.12) | 305 (0.108) | HOMO → LUMO (82%) |
| Interaction | CP | |||||||
|---|---|---|---|---|---|---|---|---|
| C7–H7···F2 | CP1 | 2.540 | 0.0058 | 0.0231 | 0.0037 | 0.0048 | 0.0010 | 0.77 |
| C3′–H3′A···F2 | CP2 | 2.669 | 0.0067 | 0.0295 | 0.0048 | 0.0061 | 0.0013 | 0.79 |
| C8–H8···F1 | CP3 | 2.731 | 0.0035 | 0.0151 | 0.0024 | 0.0031 | 0.0007 | 0.77 |
| C2′–H···O2 | CP4 | 2.724 | 0.0043 | 0.0156 | 0.0025 | 0.0032 | 0.0007 | 0.78 |
| C5–H5···N1 | CP5 | 2.779 | 0.0056 | 0.0165 | 0.0029 | 0.0035 | 0.0006 | 0.83 |
| C2′–F1···N2 | CP6 | 3.076 | 0.0058 | 0.0247 | 0.0041 | 0.0052 | 0.0010 | 0.79 |
| C2′–F2···N3 | CP7 | 3.083 | 0.0057 | 0.0236 | 0.0040 | 0.0049 | 0.0010 | 0.81 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Narváez-Ordoñez, E.G.; Pabón-Carcelén, K.A.; Zurita-Saltos, D.A.; Bonilla-Valladares, P.M.; Yánez-Darquea, T.G.; Ramos-Guerrero, L.A.; Ulic, S.E.; Jios, J.L.; Echeverría, G.A.; Piro, O.E.; et al. Synthesis, Experimental and Theoretical Study of Azidochromones. Molecules 2022, 27, 2636. https://doi.org/10.3390/molecules27092636
Narváez-Ordoñez EG, Pabón-Carcelén KA, Zurita-Saltos DA, Bonilla-Valladares PM, Yánez-Darquea TG, Ramos-Guerrero LA, Ulic SE, Jios JL, Echeverría GA, Piro OE, et al. Synthesis, Experimental and Theoretical Study of Azidochromones. Molecules. 2022; 27(9):2636. https://doi.org/10.3390/molecules27092636
Chicago/Turabian StyleNarváez-Ordoñez, Ena G., Kevin A. Pabón-Carcelén, Daniel A. Zurita-Saltos, Pablo M. Bonilla-Valladares, Trosky G. Yánez-Darquea, Luis A. Ramos-Guerrero, Sonia E. Ulic, Jorge L. Jios, Gustavo A. Echeverría, Oscar E. Piro, and et al. 2022. "Synthesis, Experimental and Theoretical Study of Azidochromones" Molecules 27, no. 9: 2636. https://doi.org/10.3390/molecules27092636
APA StyleNarváez-Ordoñez, E. G., Pabón-Carcelén, K. A., Zurita-Saltos, D. A., Bonilla-Valladares, P. M., Yánez-Darquea, T. G., Ramos-Guerrero, L. A., Ulic, S. E., Jios, J. L., Echeverría, G. A., Piro, O. E., Langer, P., Alcívar-León, C. D., & Heredia-Moya, J. (2022). Synthesis, Experimental and Theoretical Study of Azidochromones. Molecules, 27(9), 2636. https://doi.org/10.3390/molecules27092636