
Potential of Ferritin-Based Platforms for Tumor Immunotherapy

Abstract
:1. Introduction
2. Ferritin-Based Nanoplatforms for Cancer Immunotherapy
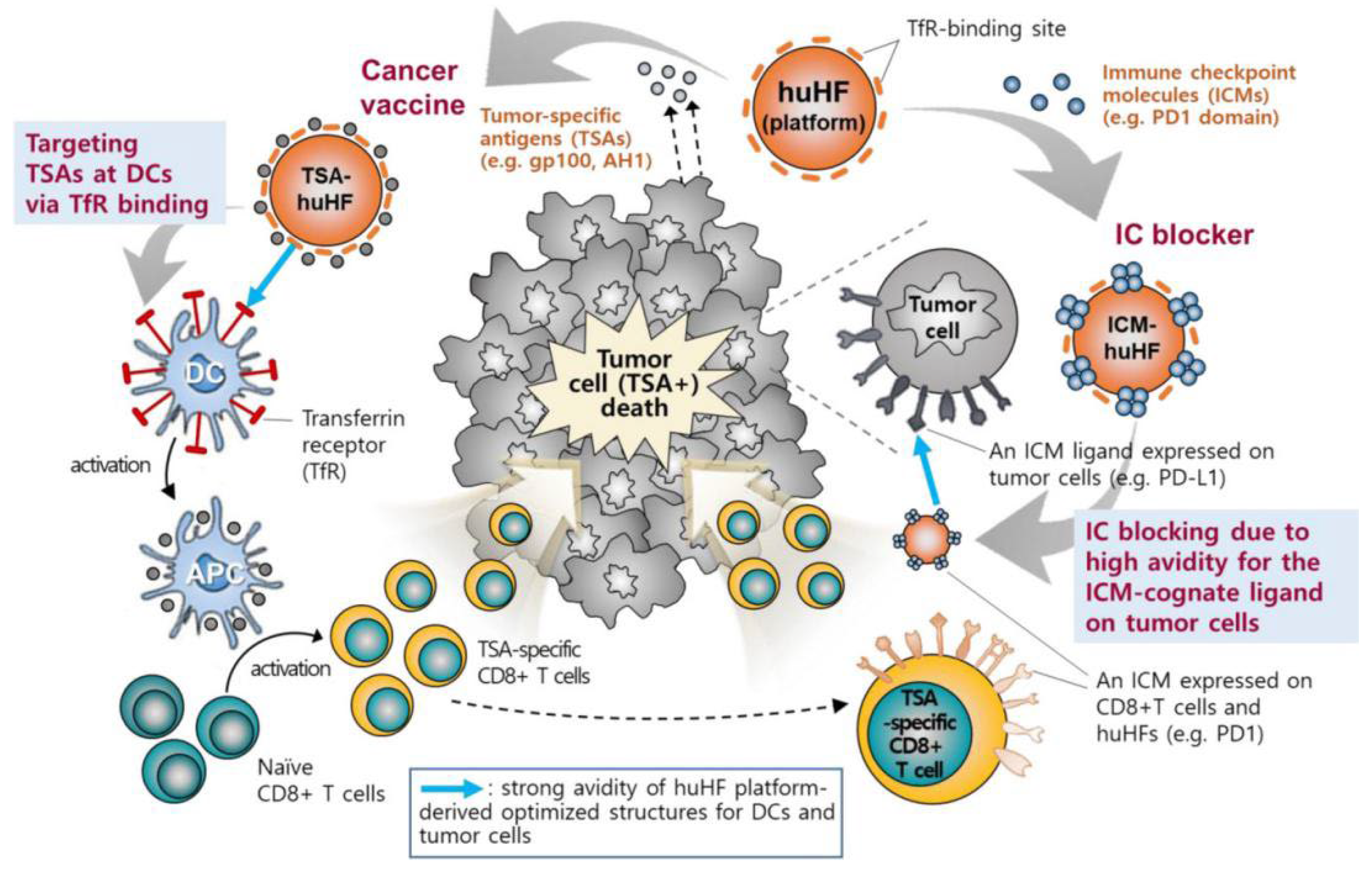
2.1. Targeting DCs
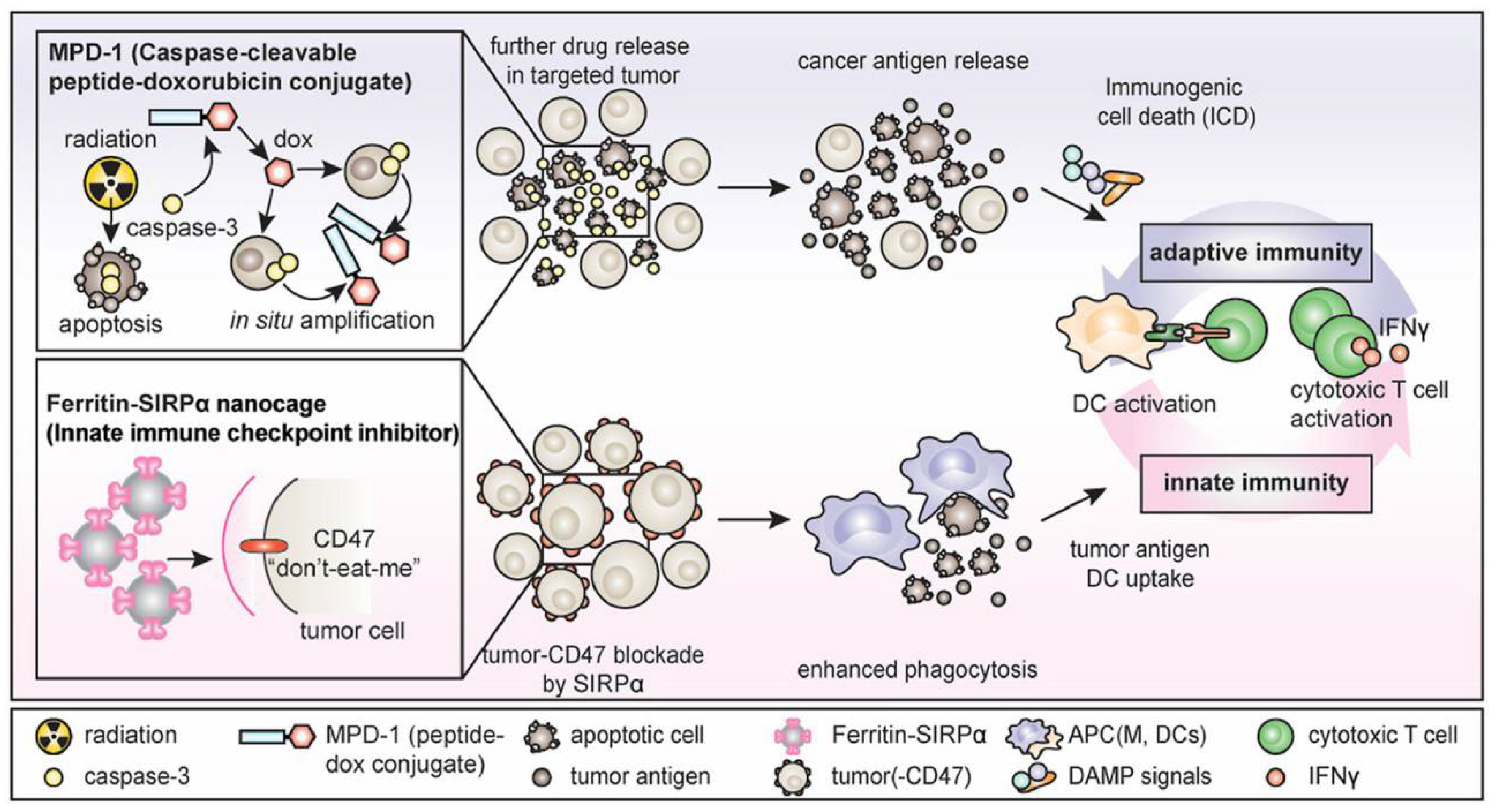
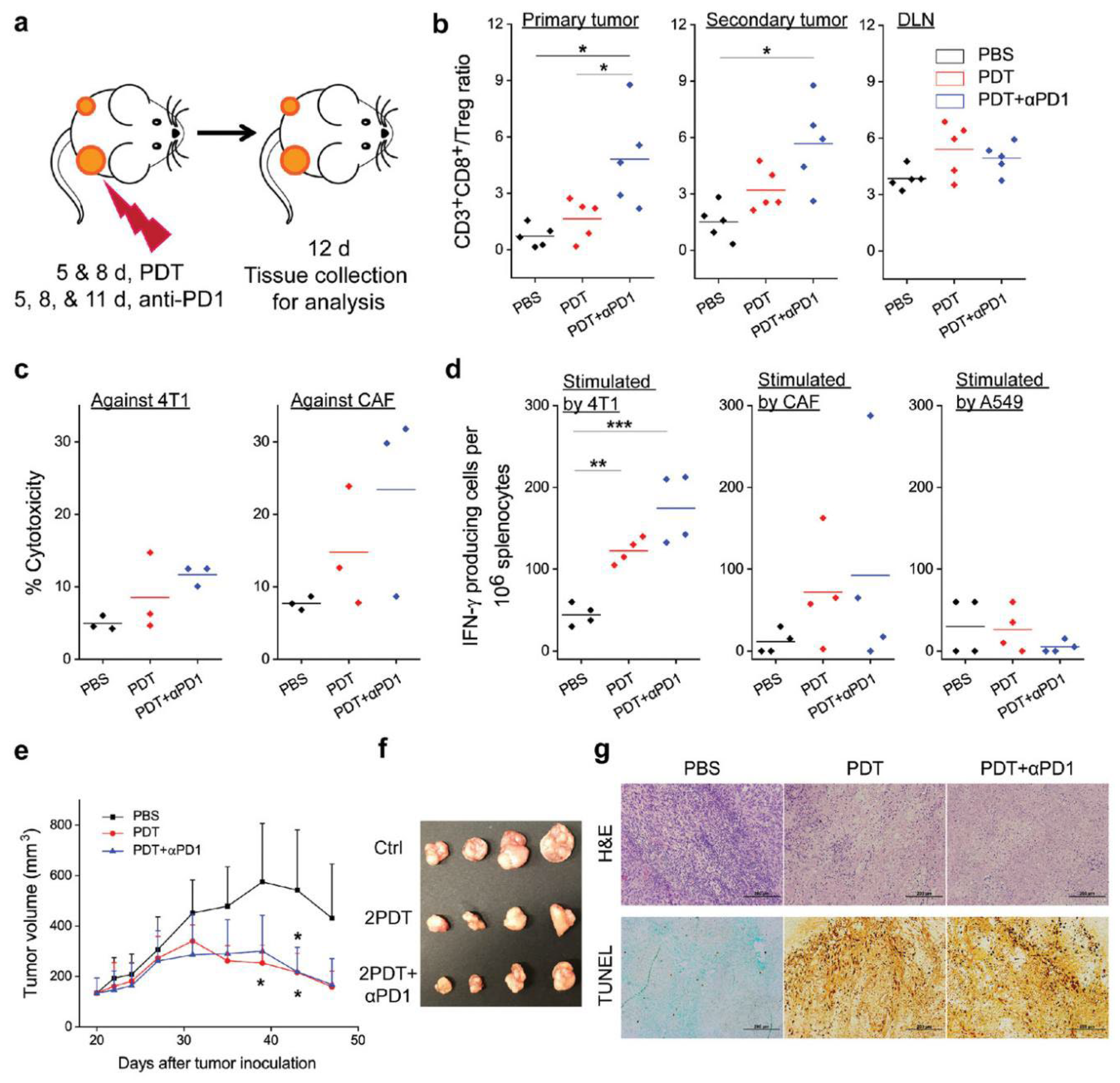
2.2. Targeting Tumor Cells
2.3. Targeting Tumor-Associated Fibroblasts
2.4. Targeting M2 TAMs
2.5. Other Applications
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gray, C.P.; Arosio, P.; Hersey, P. Association of increased levels of heavy-chain ferritin with increased CD4+ CD25+ regulatory T-cell levels in patients with melanoma. Clin. Cancer Res. 2003, 9, 2551–2559. [Google Scholar] [CrossRef] [PubMed]
- Baldi, A.; Lombardi, D.; Russo, P.; Palescandolo, E.; Luca, A.D.; Santini, D.; Baldi, F.; Rossiello, L.; Dell’Anna, M.L.; Mastrofrancesco, A.; et al. Ferritin contributes to melanoma progression by modulating cell growth and sensitivity to oxidative stress. Clin. Cancer Res. 2005, 11, 3175–3183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhushan, B.; Kumar, S.U.; Matai, I.; Sachdev, A.; Dubey, P.; Gopinath, P. Ferritin nanocages: A novel platform for biomedical applications. J. Biomed. Nanotechnol. 2014, 10, 2950–2976. [Google Scholar] [CrossRef] [PubMed]
- Veroniaina, H.; Pan, X.; Wu, Z.; Qi, X. Apoferritin: A potential nanocarrier for cancer imaging and drug delivery. Expert Rev. Anticancer. Ther. 2021, 21, 901–913. [Google Scholar] [CrossRef] [PubMed]
- Cermák, J.; Neuwirt, J. A half century since the isolation of crystalline ferritin by Professor Laufberger. Vnitr. Lek. 1986, 32, 833–835. [Google Scholar]
- Bellini, M.; Mazzucchelli, S.; Galbiati, E.; Sommaruga, S.; Fiandra, L.; Truffi, M.; Rizzuto, M.A.; Colombo, M.; Tortora, P.; Corsi, F.; et al. Protein nanocages for self-triggered nuclear delivery of DNA-targeted chemotherapeutics in cancer cells. J. Control. Release 2014, 196, 184–196. [Google Scholar] [CrossRef]
- Theil, E.C. Ferritin: Structure, gene regulation, and cellular function in animals, plants, and microorganisms. Annu. Rev. Biochem. 1987, 56, 289–315. [Google Scholar] [CrossRef]
- Song, N.; Zhang, J.; Zhai, J.; Hong, J.; Yuan, C.; Liang, M. Ferritin: A multifunctional nanoplatform for biological detection, imaging diagnosis, and drug delivery. Acc. Chem. Res. 2021, 54, 3313–3325. [Google Scholar] [CrossRef]
- Xue, L.; Deng, D.; Sun, J. Magnetoferritin: Process, prospects, and their biomedical applications. Int. J. Mol. Sci. 2019, 20, 2426. [Google Scholar] [CrossRef] [Green Version]
- He, J.; Fan, K.; Yan, X. Ferritin drug carrier (FDC) for tumor targeting therapy. J. Control. Release 2019, 311–312, 288–300. [Google Scholar] [CrossRef]
- Zhao, G.; Arosio, P.; Chasteen, N.D. Iron(II) and hydrogen peroxide detoxification by human H-chain ferritin. An EPR spin-trapping study. Biochemistry 2006, 45, 3429–3436. [Google Scholar] [CrossRef] [PubMed]
- Chasteen, N.D.; Harrison, P.M. Mineralization in ferritin: An efficient means of iron storage. J. Struct. Biol. 1999, 126, 182–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zámocký, M.; Koller, F. Understanding the structure and function of catalases: Clues from molecular evolution and in vitro mutagenesis. Prog. Biophys. Mol. Biol. 1999, 72, 19–66. [Google Scholar] [CrossRef]
- Sun, X.; Hong, Y.; Gong, Y.; Zheng, S.; Xie, D. Bioengineered ferritin nanocarriers for cancer therapy. Int. J. Mol. Sci. 2021, 22, 7023. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Cheng, D.; He, J.; Hong, J.; Yuan, C.; Liang, M. Cargo loading within ferritin nanocages in preparation for tumor-targeted delivery. Nat. Protoc. 2021, 16, 4878–4896. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.C. Iron storage in bacteria. Adv. Microb. Physiol. 1998, 40, 281–351. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, I.; Iwahori, K.; Kumagai, S. Ferritin in the field of nanodevices. Biochim. Biophys. Acta 2010, 1800, 846–857. [Google Scholar] [CrossRef]
- Levi, S.; Arosio, P. Mitochondrial ferritin. Int. J. Biochem. Cell Biol. 2004, 36, 1887–1889. [Google Scholar] [CrossRef]
- Arosio, P.; Levi, S. Ferritin, iron homeostasis, and oxidative damage. Free. Radic. Biol. Med. 2002, 33, 457–463. [Google Scholar] [CrossRef]
- Everett, J.; Brooks, J.; Lermyte, F.; O’Connor, P.B.; Sadler, P.J.; Dobson, J.; Collingwood, J.F.; Telling, N.D. IRON stored in ferritin is chemically reduced in the presence of aggregating Aβ(1-42). Sci. Rep. 2020, 10, 10332. [Google Scholar] [CrossRef]
- Sato, D.; Ikeguchi, M. Mechanisms of ferritin assembly studied by time-resolved small-angle X-ray scattering. Biophys. Rev. 2019, 11, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Simsek, E.; Kilic, M.A. Magic ferritin: A novel chemotherapeutic encapsulation bullet. J. Magn. Magn. Mater. 2005, 293, 509–513. [Google Scholar] [CrossRef]
- Bou-Abdallah, F. The iron redox and hydrolysis chemistry of the ferritins. Biochim. Biophys. Acta 2010, 1800, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, X.; Hong, J.; Tang, A.; Liu, Y.; Xie, N.; Nie, G.; Yan, X.; Liang, M. Biochemistry of mammalian ferritins in the regulation of cellular iron homeostasis and oxidative responses. Sci. China Life Sci. 2021, 64, 352–362. [Google Scholar] [CrossRef]
- Mainini, F.; Bonizzi, A.; Sevieri, M.; Sitia, L.; Truffi, M.; Corsi, F.; Mazzucchelli, S. Protein-based nanoparticles for the imaging and treatment of solid tumors: The case of ferritin nanocages, a narrative review. Pharmaceutics 2021, 13, 2000. [Google Scholar] [CrossRef]
- Fan, K.; Gao, L.; Yan, X. Human ferritin for tumor detection and therapy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2013, 5, 287–298. [Google Scholar] [CrossRef]
- Gomhor, J.; Alqaraghuli, H.; Kashanian, S.; Rafipour, R.; Mahdavian, E.; Mansouri, K. Development and characterization of folic acid-functionalized apoferritin as a delivery vehicle for epirubicin against MCF-7 breast cancer cells. Artif. Cells Nanomed. Biotechnol. 2018, 46 (Suppl. 3), S847–S854. [Google Scholar] [CrossRef]
- Tesarova, B.; Musilek, K.; Rex, S.; Heger, Z. Taking advantage of cellular uptake of ferritin nanocages for targeted drug delivery. J. Control. Release 2020, 325, 176–190. [Google Scholar] [CrossRef]
- Cheng, X.J.; Fan, K.; Wang, L.; Ying, X.J.; Sanders, A.J.; Guo, T.; Xing, X.F.; Zhou, M.; Du, H.; Hu, Y.; et al. TfR1 binding with H-ferritin nanocarrier achieves prognostic diagnosis and enhances the therapeutic efficacy in clinical gastric cancer. Cell Death Dis. 2020, 11, 92. [Google Scholar] [CrossRef] [Green Version]
- Callens, C.; Moura, I.C.; Lepelletier, Y.; Coulon, S.; Renand, A.; Dussiot, M.; Ghez, D.; Benhamou, M.; Monteiro, R.C.; Bazarbachi, A.; et al. Recent advances in adult T-cell leukemia therapy: Focus on a new anti-transferrin receptor monoclonal antibody. Leukemia 2008, 22, 42–48. [Google Scholar] [CrossRef]
- Fan, K.L.; Jia, X.H.; Zhou, M.; Wang, K.; Conde, J.; He, J.Y.; Tian, J.; Yan, X.Y. Ferritin nanocarrier traverses the blood brain barrier and kills glioma. ACS Nano 2018, 12, 4105–4115. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Zhang, S.; Yang, Y.; Cheng, L.; Li, C.; Dai, L.; Zhang, X.; Fan, P.; Tian, H.; Wang, R.; et al. Therapeutic upregulation of Class A scavenger receptor member 5 inhibits tumor growth and metastasis. Cancer Sci. 2012, 103, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Paragas, N.; Ned, R.M.; Qiu, A.; Viltard, M.; Leete, T.; Drexler, I.R.; Chen, X.; Sanna-Cherchi, S.; Mohammed, F.; et al. Scara5 is a ferritin receptor mediating non-transferrin iron delivery. Dev. Cell 2009, 16, 35–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wetz, K.; Crichton, R.R. Chemical modification as a probe of the topography and reactivity of horse-spleen apoferritin. Eur. J. Biochem. 1976, 61, 545–550. [Google Scholar] [CrossRef] [PubMed]
- Martsev, S.P.; Vlasov, A.P.; Arosio, P. Distinct stability of recombinant L and H subunits of human ferritin: Calorimetric and ANS binding studies. Protein Eng. Des. Sel. 1998, 11, 377–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Li, J.; Gu, P.; Fan, X. The application of nanoparticles in cancer immunotherapy: Targeting tumor microenvironment. Bioact. Mater. 2020, 6, 1973–1987. [Google Scholar] [CrossRef]
- Fu, C.; Zhou, L.; Mi, Q.S.; Jiang, A. Plasmacytoid dendritic cells and cancer immunotherapy. Cells. 2022, 11, 222. [Google Scholar] [CrossRef]
- Abakushina, E.V.; Popova, L.I.; Zamyatnin, A.A., Jr.; Werner, J.; Mikhailovsky, N.V.; Bazhin, A.V. The advantages and challenges of anticancer dendritic cell vaccines and NK cells in adoptive cell immunotherapy. Vaccines 2021, 9, 1363. [Google Scholar] [CrossRef]
- Shaw, C.A.; Tomljenovic, L. Aluminum in the central nervous system (CNS): Toxicity in humans and animals, vaccine adjuvants, and autoimmunity. Immunol. Res. 2013, 56, 304–316. [Google Scholar] [CrossRef]
- Lee, B.R.; Ko, H.K.; Ryu, J.H.; Ahn, K.Y.; Lee, Y.-H.; Oh, S.J.; Na, J.H.; Kim, T.W.; Byun, Y.; Kwon, I.K.; et al. Engineered human ferritin nanoparticles for direct delivery of tumor antigens to lymph node and cancer immunotherapy. Sci. Rep. 2016, 6, 35182. [Google Scholar] [CrossRef] [Green Version]
- Lee, B.-R.; Lee, H.-J.; Huh, J.; Yoon, C.J.; Oh, S.J.; Song, K.-H.; Jeong, S.; Kim, J.; Lee, K.-M.; Shin, B.S.; et al. Human ferritin platform and its optimized structures to enhance anti-cancer immunity. Adv. Therap. 2021, 4, 2000208. [Google Scholar] [CrossRef]
- Han, J.A.; Kang, Y.J.; Shin, C.; Ra, J.-S.; Shin, H.-H.; Hong, S.Y.; Do, Y.; Kang, S. Ferritin protein cage nanoparticles as versatile antigen delivery nanoplatforms for dendritic cell (DC)-based vaccine development. Nanomedicine 2014, 10, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Liu, Z.; Zhou, X.; Guo, Z.; Zhang, J.; Zhu, P.; Yao, S.; Zhu, M. Ferritin nanoparticle-based SpyTag/SpyCatcher-enabled click vaccine for tumor immunotherapy. Nanomedicine 2019, 16, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Mariz, F.C.; Zhao, X.; Spagnoli, G.; Ottonello, S.; Müller, M. Broad neutralization responses against oncogenic human papillomaviruses induced by a minor capsid l2 polytope genetically incorporated into bacterial ferritin nanoparticles. Front Immunol. 2020, 11, 606569. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, S.; Guo, C. Crosstalk between macrophages and natural killer cells in the tumor microenvironment. Int. Immunopharmacol. 2021, 101 Pt B, 108374. [Google Scholar] [CrossRef]
- Zhang, J.; Jin, S.; Guo, X.; Qian, W. Targeting the CD47-SIRPα signaling axis: Current studies on B-cell lymphoma immunotherapy. J. Int. Med. Res. 2018, 46, 4418–4426. [Google Scholar] [CrossRef] [Green Version]
- Lee, E.J.; Nam, G.H.; Lee, N.K.; Kih, M.; Koh, E.; Kim, Y.K.; Hong, Y.; Kim, S.; Park, S.-Y.; Jeong, C.; et al. Nanocage-therapeutics prevailing phagocytosis and immunogenic cell death awakens immunity against cancer. Adv. Mater. 2018, 30, 1705581. [Google Scholar] [CrossRef]
- Cho, E.; Nam, G.H.; Hong, Y.; Cho, E.; Nam, G.-H.; Hong, Y.; Kim, Y.K.; Kim, D.-H.; Yang, Y.; Kim, I.-S. Comparison of exosomes and ferritin protein nanocages for the delivery of membrane protein therapeutics. J. Control. Release 2018, 279, 326–335. [Google Scholar] [CrossRef]
- Lee, N.K.; Choi, J.U.; Kim, H.R.; Chung, S.W.; Ko, Y.G.; Cho, Y.S.; Park, S.J.; Lee, E.J.; Kim, S.Y.; Kim, I.-S.; et al. Caspase-cleavable peptide-doxorubicin conjugate in combination with CD47-antagonizing nanocage therapeutics for immune-mediated elimination of colorectal cancer. Biomaterials 2021, 277, 121105. [Google Scholar] [CrossRef]
- Jeon, I.S.; Yoo, J.D.; Gurung, S.; Kim, M.; Lee, C.; Park, E.J.; Park, R.-W.; Lee, B.; Kim, S. Anticancer nanocage platforms for combined immunotherapy designed to harness immune checkpoints and deliver anticancer drugs. Biomaterials 2021, 270, 120685. [Google Scholar] [CrossRef]
- Zhen, Z.; Tang, W.; Wang, M. Protein nanocage mediated fibroblast-activation protein targeted photoimmunotherapy to enhance cytotoxic t cell infiltration and tumor control. Nano Lett. 2017, 17, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Zhen, Z.; Paschall, A.V.; Xue, L.; Yang, X.; Blackwell, A.-G.B.; Cao, Z.; Zhang, W.; Wang, M.; Teng, Y.; et al. FAP-targeted photodynamic therapy mediated by ferritin nanoparticles elicits an immune response against cancer cells and cancer associated fibroblasts. Adv. Funct. Mater. 2021, 31, 2007017. [Google Scholar] [CrossRef]
- Shi, L.; Gu, H. Emerging nanoparticle strategies for modulating tumor-associated macrophage polarization. Biomolecules 2021, 11, 1912. [Google Scholar] [CrossRef]
- Xu, T.; Yu, S.; Zhang, J.; Wu, S. Dysregulated tumor-associated macrophages in carcinogenesis, progression and targeted therapy of gynecological and breast cancers. J. Hematol. Oncol. 2021, 14, 181. [Google Scholar] [CrossRef] [PubMed]
- Shan, H.; Dou, W.; Zhang, Y.; Qi, M. Targeted ferritin nanoparticle encapsulating CpG oligodeoxynucleotides induces tumor-associated macrophage M2 phenotype polarization into M1 phenotype and inhibits tumor growth. Nanoscale 2020, 12, 22268–22280. [Google Scholar] [CrossRef]
- Eissa, I.R.; Mukoyama, N.; Abdelmoneim, M.; Abdelmoneim, M.; Naoe, Y.; Matsumura, S.; Bustos-Villalobos, I.; Ichinose, T.; Miyajima, N.; Morimoto, D.; et al. Oncolytic herpes simplex virus HF10 (canerpaturev) promotes accumulation of CD8+ PD-1- tumor-infiltrating T cells in PD-L1-enriched tumor microenvironment. Int. J. Cancer 2021, 149, 214–227. [Google Scholar] [CrossRef]
- Zhulai, G.; Oleinik, E. Targeting regulatory T cells in anti-PD-1/PD-L1 cancer immunotherapy. Scand. J. Immunol. 2022, 95, e13129. [Google Scholar] [CrossRef]
- Xie, W.; Medeiros, L.J.; Li, S.; Yin, C.C.; Khoury, J.D.; Xu, J. PD-1/PD-L1 pathway and its blockade in patients with classic hodgkin lymphoma and non-hodgkin large-cell lymphomas. Curr. Hematol. Malig. Rep. 2020, 15, 372–381. [Google Scholar] [CrossRef]
- Kim, G.B.; Sung, H.D.; Nam, G.H.; Kim, w.; Kim, S.; Kang, D.; Lee, E.J.; Kim, I.-S. Design of PD-1-decorated nanocages targeting tumor-draining lymph node for promoting T cell activation. J. Control. Release 2021, 333, 328–338. [Google Scholar] [CrossRef]
- Kim, M.; Tomek, P. Tryptophan: A rheostat of cancer immune escape mediated by immunosuppressive enzymes IDO1 and TDO. Front. Immunol. 2021, 12, 636081. [Google Scholar] [CrossRef]
- Liu, X.; Newton, R.C.; Friedman, S.M.; Scherle, P.A. Indoleamine 2,3-dioxygenase, an emerging target for anti-cancer therapy. Curr. Cancer Drug Targets 2009, 9, 938–952. [Google Scholar] [CrossRef] [PubMed]
- Ricciuti, B.; Leonardi, G.C.; Puccetti, P.; Fallarino, F.; Bianconi, V.; Sahebkar, A.; Baglivo, S.; Chiari, R.; Pirro, M. Targeting indoleamine-2,3-dioxygenase in cancer: Scientific rationale and clinical evidence. Pharmacol. Ther. 2019, 196, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Komiya, T.; Huang, C.H. Updates in the clinical development of epacadostat and other indoleamine 2,3-dioxygenase 1 inhibitors (IDO1) for human cancers. Front. Oncol. 2018, 8, 423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Zhang, W.; Jiang, W.; Kumar, A.; Zhou, S.; Cao, Z.; Zhan, S.; Yang, W.; Liu, R.; Teng, T.; et al. Nanoconjugates to enhance PDT-mediated cancer immunotherapy by targeting the indoleamine-2,3-dioxygenase pathway. J. Nanobiotechnol. 2021, 19, 182. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Park, J.E.; Im, E.; Cho, Y.; Lee, J.; Lee, H.-J.; Sim, D.-Y.; Park, W.-Y.; Shim, B.-S.; Kim, S.-H. Recent advances in nanotechnology with nano-phytochemicals: Molecular mechanisms and clinical implications in cancer progression. Int. J. Mol. Sci. 2021, 22, 3571. [Google Scholar] [CrossRef]
- Del Prado-Audelo, M.L.; Caballero-Florán, I.H.; Meza-Toledo, J.A.; Mendoza-Muñoz, N.; González-Torres, N.; Florán, B.; Cortés, H.; Leyva-Gómez, G. Formulations of curcumin nanoparticles for brain diseases. Biomolecules 2019, 9, 56. [Google Scholar] [CrossRef] [Green Version]
- Moutabian, H.; Ghahramani-Asl, R.; Mortezazadeh, T.; Laripour, R.; Narmani, A.; Zamani, H.; Ataei, G.; Bagheri, H.; Farhood, B.; Sathyapalan, T.; et al. The cardioprotective effects of nano-curcumin against doxorubicin-induced cardiotoxicity: A systematic review. Biofactors 2022, 10. [Google Scholar] [CrossRef]
- Ibrahim, M.; Abuwatfa, W.H.; Awad, N.S.; Sabouni, R.; Husseini, G.A. Encapsulation, release, and cytotoxicity of doxorubicin loaded in liposomes, micelles, and metal-organic frameworks: A review. Pharmaceutics 2022, 14, 254. [Google Scholar] [CrossRef]
- Jain, A.; Tiwari, A.; Verma, A.; Saraf, S.; Jain, S.K. Combination cancer therapy using multifunctional liposomes. Crit. Rev. Ther. Drug Carr. Syst. 2020, 37, 105–134. [Google Scholar] [CrossRef]
- Rui, M.; Xin, Y.; Li, R.; Ge, Y.; Feng, C.; Xu, X. Targeted biomimetic nanoparticles for synergistic combination chemotherapy of paclitaxel and doxorubicin. Mol. Pharm. 2017, 14, 107–123. [Google Scholar] [CrossRef]
- Feng, C.; Zhang, H.; Chen, J.; Wang, S.; Xin, Y.; Qu, Y.; Zhang, Q.; Ji, W.; Yamashita, F.; Rui, M.; et al. Ratiometric co-encapsulation and co-delivery of doxorubicin and paclitaxel by tumor-targeted lipodisks for combination therapy of breast cancer. Int. J. Pharm. 2019, 560, 191–204. [Google Scholar] [CrossRef] [PubMed]
- Benameur, T.; Soleti, R.; Porro, C. The potential neuroprotective role of free and encapsulated quercetin mediated by miRNA against neurological diseases. Nutrients. 2021, 13, 1318. [Google Scholar] [CrossRef]
- Fan, K.; Cao, C.; Pan, Y.; Lu, D.; Yang, D.; Feng, J.; Song, L.; Liang, M.; Yan, X. Magnetoferritin nanoparticles for targeting and visualizing tumour tissues. Nat. Nanotechnol. 2012, 7, 459–464. [Google Scholar] [CrossRef]
- Zhao, Y.; Liang, M.; Li, X.; Fan, K.; Xiao, J.; Li, Y.; Shi, H.; Wang, F.; Choi, H.S.; Cheng, D.; et al. Bioengineered magnetoferritin nanoprobes for single-dose nuclear-magnetic resonance tumor imaging. ACS Nano 2016, 10, 4184–4191. [Google Scholar] [CrossRef] [PubMed]
- Bowman, A.W.; Gooch, C.R.; Alexander, L.F.; Desai, M.A.; Bolan, C.W. Vascular applications of ferumoxytol-enhanced magnetic resonance imaging of the abdomen and pelvis. Abdom. Radiol. 2021, 46, 2203–2218. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Huang, P.; Jacobson, O.; Wang, Z.; Liu, Y.; Lin, L.; Lin, J.; Lu, N.; Zhang, H.; Tian, R.; et al. Biomineralization-inspired synthesis of copper sulfide-ferritin nanocages as cancer theranostics. ACS Nano 2016, 10, 3453–3460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nieves, L.M.; Mossburg, K.; Hsu, J.C.; Maidment, A.D.A.; Cormode, D.P. Silver chalcogenide nanoparticles: A review of their biomedical applications. Nanoscale 2021, 13, 19306–19323. [Google Scholar] [CrossRef]
- Shen, Y.; Lifante, J.; Ximendes, E.; Santos, H.D.A.; Ruiz, D.; Juárez, B.H.; Gutiérrez, I.Z.; Vera, V.T.; Retama, J.R.; Rodríguez, E.M.; et al. Perspectives for Ag2S NIR-II nanoparticles in biomedicine: From imaging to multifunctionality. Nanoscale 2019, 11, 19251–19264. [Google Scholar] [CrossRef]
- Palombarini, F.; Masciarelli, S.; Incocciati, A.; Liccardo, F.; Fabio, E.D.; Iazzetti, A.; Fabrizi, G.; Fazi, F.; Macone, A.; Bonamore, A.; et al. Self-assembling ferritin-dendrimer nanoparticles for targeted delivery of nucleic acids to myeloid leukemia cells. J. Nanobiotechnol. 2021, 19, 172. [Google Scholar] [CrossRef]
- Rodrigues, M.Q.; Alves, P.M.; Roldão, A. Functionalizing ferritin nanoparticles for vaccine development. Pharmaceutics 2021, 13, 1621. [Google Scholar] [CrossRef]
- Joyce, M.G.; King, H.A.D.; Elakhal-Naouar, I.; Ahmed, A.; Peachman, K.K.; Cincotta, C.M.; Subra, C.; Chen, R.E.; Thomas, P.V.; Chen, W.-H.; et al. A SARS-CoV-2 ferritin nanoparticle vaccine elicits protective immune responses in nonhuman primates. Sci. Transl. Med. 2021, 6, 151. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Rho, Y.; Jin, K.S.; Ahn, B.; Jung, S.; Kim, H.; Ree, M. pH-dependent structures of ferritin and apoferritin in solution: Disassembly and reassembly. Biomacromolecules 2011, 12, 1629–1640. [Google Scholar] [CrossRef] [PubMed]
- Theil, E.C. Ferritin protein nanocages use ion channels, catalytic sites, and nucleation channels to manage iron/oxygen chemistry. Curr. Opin. Chem. Biol. 2011, 15, 304–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Högbom, M. Metal use in ribonucleotide reductase R2, di-iron, di-manganese and heterodinuclear--an intricate bioinorganic workaround to use different metals for the same reaction. Metallomics 2011, 3, 110–120. [Google Scholar] [CrossRef] [Green Version]
- Silva, F.; Sitia, L.; Allevi, R.; Bonizzi, A.; Sevieri, M.; Morasso, C.; Truffi, M.; Corsi, F.; Mazzucchelli, S. Combined method to remove endotoxins from protein nanocages for drug delivery applications: The case of human ferritin. Pharmaceutics 2021, 13, 229. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Advantage | Reasons |
|---|---|
| Good biocompatibility | Ferritin is an iron storage protein that widely found in various living organisms (plants, amphibians, mammals, and others) [23]. The composition of ferritin in mammals is similar and includes an H chain and an L chain [24]. Therefore, ferritin has a high biocompatibility based on its endogenous homology [25]. Using ferritin as a carrier for drug delivery has almost no toxic side effects. |
| High thermal stability and acid and alkali resistance for easy production | Protein purification often requires complex procedures to separate the target protein from other host cell proteins. Ferritin can withstand a wide range of pH values and temperatures as high as 75 °C for 10 min while still maintaining its icosahedral structure [26]. Therefore, the isolation of ferritin can be carried out using a simple, one-step heat treatment that denatures more than 80% of host cell proteins [27,28]. The excellent physical and chemical properties of ferritin greatly simplify the production process, requiring no special handling during transportation and storage, which distinguishes this protein from many other protein-based drug delivery vehicles and contributes to its clinical translation. |
| Natural cell-targeting ability | The residues Q14, D15, E17–A19, N21, and R22 in the N-terminal region of the A helix of the H-chain subunit can interact with several residues (R79, F81, Q83, K86, and K87) in the BC loop (short-loop region between the B and C helices) to achieve specific binding of ferritin to the transferrin receptor (TfR) [29].TfR1 is an important transmembrane glycoprotein that regulates cell growth and is expressed on the surface of endothelial cells, erythrocytes, and other cells [10]; however, it is overexpressed in proliferating cells, which require more iron. Thus, TfR1 is widely overexpressed in antigen-presenting cells (APCs) in many malignant tumors, as well as tumor cells. The density of TfR1 on the surface of proliferating cells is approximately 100-fold higher than that in nonproliferating cells. When the density of TfR1 on the cell surface is high, the formed ferritin–TfR1 complex is internalized into intracellular lysosomes for targeted delivery [30]. Thus, H-chain-enriched ferritin is able to target a broad range of tumors, with 10-fold greater potency than that due to the enhanced permeability and retention effect alone, providing a pathway for drug delivery and diagnostic therapy [26]. When the density of TfR1 on the cell surface is low, the formed ferritin–TfR1 complex does not enter intracellular lysosomes to achieve transmembrane transport [31]. In addition, mouse and hFTns are able to interact with mouse T-cell immunoglobulin and mucin domain 2 [26].The L-chain subunit of ferritin has been shown to bind to scavenger receptor class A member 5 (SCARA5), a newly discovered class A scavenger receptor protein. SCARA5 function has not been fully defined, but its expression is closely related to tumor growth and development [32]. Ferritin from horse spleen is a commonly used natural ferritin, consisting of 90% L-chain subunits and 10% H-chain subunits. Studies have found that it can accumulate in large amounts at tumor sites, possibly due to the L chain. This subunit binds to mouse SCARA5 but not to mouse TfR1 [33]. |
| Easily modifiable surface | Each of the 24 subunits of ferritin has amino, carboxyl, sulfhydryl, and other active groups that can be modified by chemical methods, and the amino acid sequence of ferritin can be precisely modified using biological methods. A study showed that amino (3 ± 0.3 lysine residues) and carboxyl (7.1 ± 0.7) groups can be chemically modified on each subunit of the iron-rich ferritin derived from horse spleen [34]. When the iron core is removed, sulfhydryl (derived from 1.0 ± 0.1 cysteine), amino (4.4 ± 0.4 lysine residues), and carboxyl (11.0 ± 0.4) groups can be chemically modified on each subunit of horse-spleen-derived apoferritin [34]. The C-terminal and surface-loop regions of each subunit may provide sites for the insertion of various types of antigenic peptides and small protein antigens. |
| Small particle size | In normal tissue, the microvascular endothelial space is dense and structurally complete; thus, macromolecules and lipid particles cannot easily penetrate the blood vessel wall. Meanwhile, in solid tumor tissue, the structural integrity of abundant blood vessels is poor, which results in nano-sized openings between microvascular endothelial cells. The lymphatic return in solid tumors is missing. Consequently, drugs can selectively accumulate in tumors because of the enhanced permeability and retention effect. Ferritin is a nanocage with an inner diameter of 8 nm and an outer diameter of 12 nm. The appropriate particle size facilitates its entrance to the target site through the opening of inflammatory microvascular endothelial cells and deep penetration into the tissue. |
| Hydrophilic channels and the cavity that can be loaded with various drugs | The structure of ferritin, whether it is pure H-chain ferritin, pure L-chain ferritin, or mixed H- and L-chain ferritin, can be disassembled into its various subunits under extremely acidic (pH 2) or alkaline (pH 12) conditions [35]. Then, it reassembles without any external force when the ambient pH returns to the physiological range [35]. This shape-memory capability allows the encapsulation of various types of drugs in the ferritin cavity or the structure serves as a template to mineralize various metal oxides for disease diagnosis and treatment. |
| Challenge | Reasons |
|---|---|
| Standardization of drug loading | Intact hollow, spherical apoferritin is stable in the pH range of 3.40–10.0 [28,82]. As the pH is lowered from 3.40 to 0.80, apoferritin undergoes a gradual breakdown, first forming a hollow sphere with two pores, then a head-mounted structure, and finally a rod-like oligomer. As the pH is increased from 1.96, the disassembled rod-like oligomers first revert to an earphone-like structure and then to a hollow, spherical structure with two-hole defects. When pH continues to increase to neutral or slightly alkaline values, the hollow spherical structure with the two-hole defect still fails to heal [8]. This may affect the homogeneity of ferritin nanocarriers and their in vivo behavior after administration. Therefore, more reliable drug loading methods are needed to obtain defect-free ferritin formulations. |
| The modification site of ferritin [12] | Modifications at different sites in the ferritin sequence result in different display states of polypeptides or antigens, which may be stretched or gathered and may also affect the natural self-assembly ability and targeting function of ferritin [28,83,84,85]. Therefore, irrespective of whether a chemical modification or gene recombination is used to load antigens or peptides, more investigation is needed on the modification sites. |
| Animal models | At present, the animal models used to test ferritin-based tumor immune preparations are mainly mice, and the tumor microenvironment is quite different from the clinical reality. Therefore, it is necessary to develop antitumor models in larger animals to simulate actual clinical conditions, especially for the study of antitumor vaccines. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, X.; Tian, K.; Lou, X.; Du, Y. Potential of Ferritin-Based Platforms for Tumor Immunotherapy. Molecules 2022, 27, 2716. https://doi.org/10.3390/molecules27092716
Xu X, Tian K, Lou X, Du Y. Potential of Ferritin-Based Platforms for Tumor Immunotherapy. Molecules. 2022; 27(9):2716. https://doi.org/10.3390/molecules27092716
Chicago/Turabian StyleXu, Xiaoling, Kewei Tian, Xuefang Lou, and Yongzhong Du. 2022. "Potential of Ferritin-Based Platforms for Tumor Immunotherapy" Molecules 27, no. 9: 2716. https://doi.org/10.3390/molecules27092716
APA StyleXu, X., Tian, K., Lou, X., & Du, Y. (2022). Potential of Ferritin-Based Platforms for Tumor Immunotherapy. Molecules, 27(9), 2716. https://doi.org/10.3390/molecules27092716