
Development, In-Vitro Characterization and Preclinical Evaluation of Esomeprazole-Encapsulated Proniosomal Formulation for the Enhancement of Anti-Ulcer Activity


 , , , , , ,
, , , , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Method
2.2.1. Box-Behnken Statistical Design
2.2.2. Preparation of Proniosomes
2.2.3. Vesicle Size and Zeta Potential Measurement
2.2.4. Drug Entrapment Efficiency
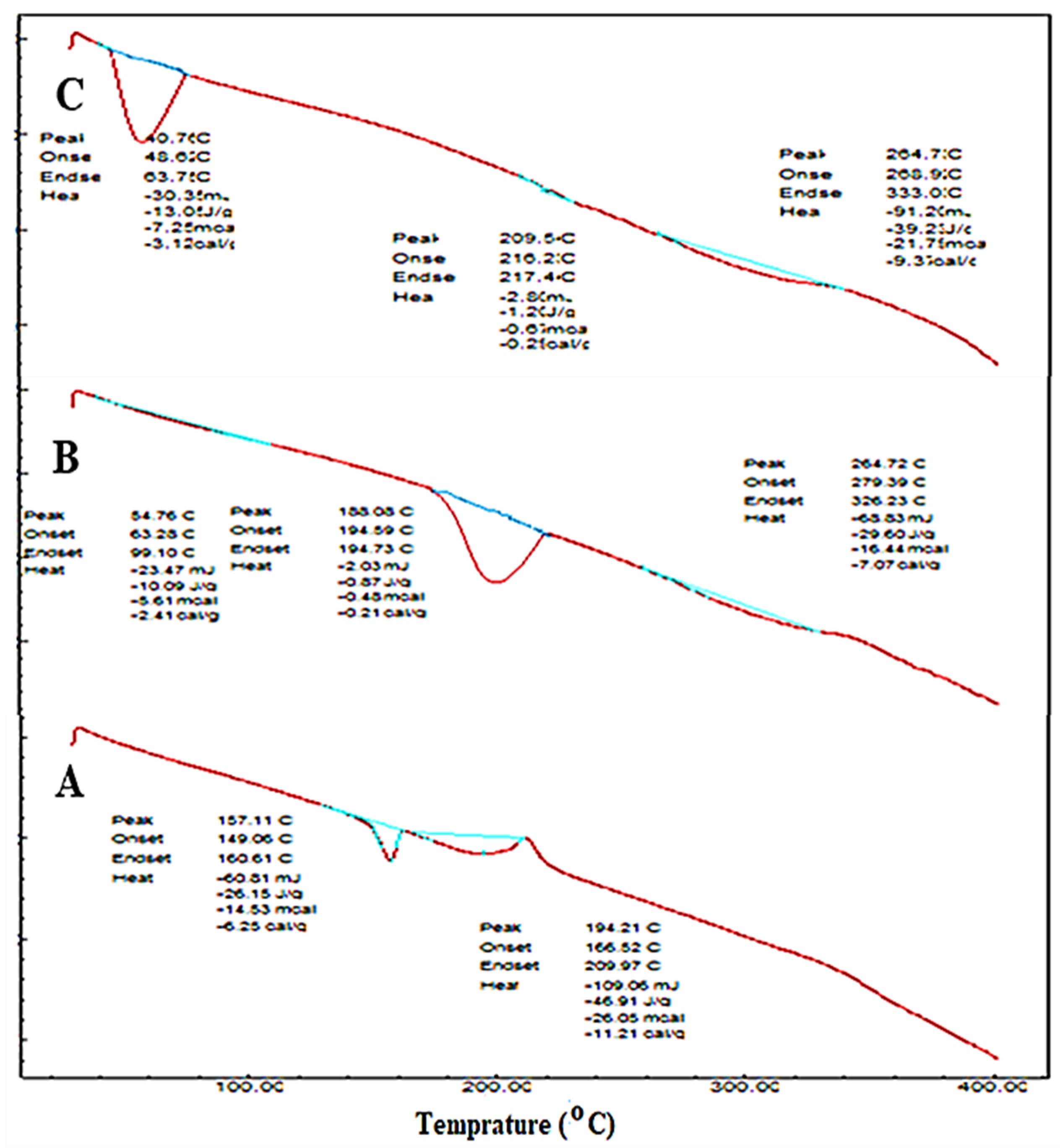
2.2.5. Thermal Transition Analysis
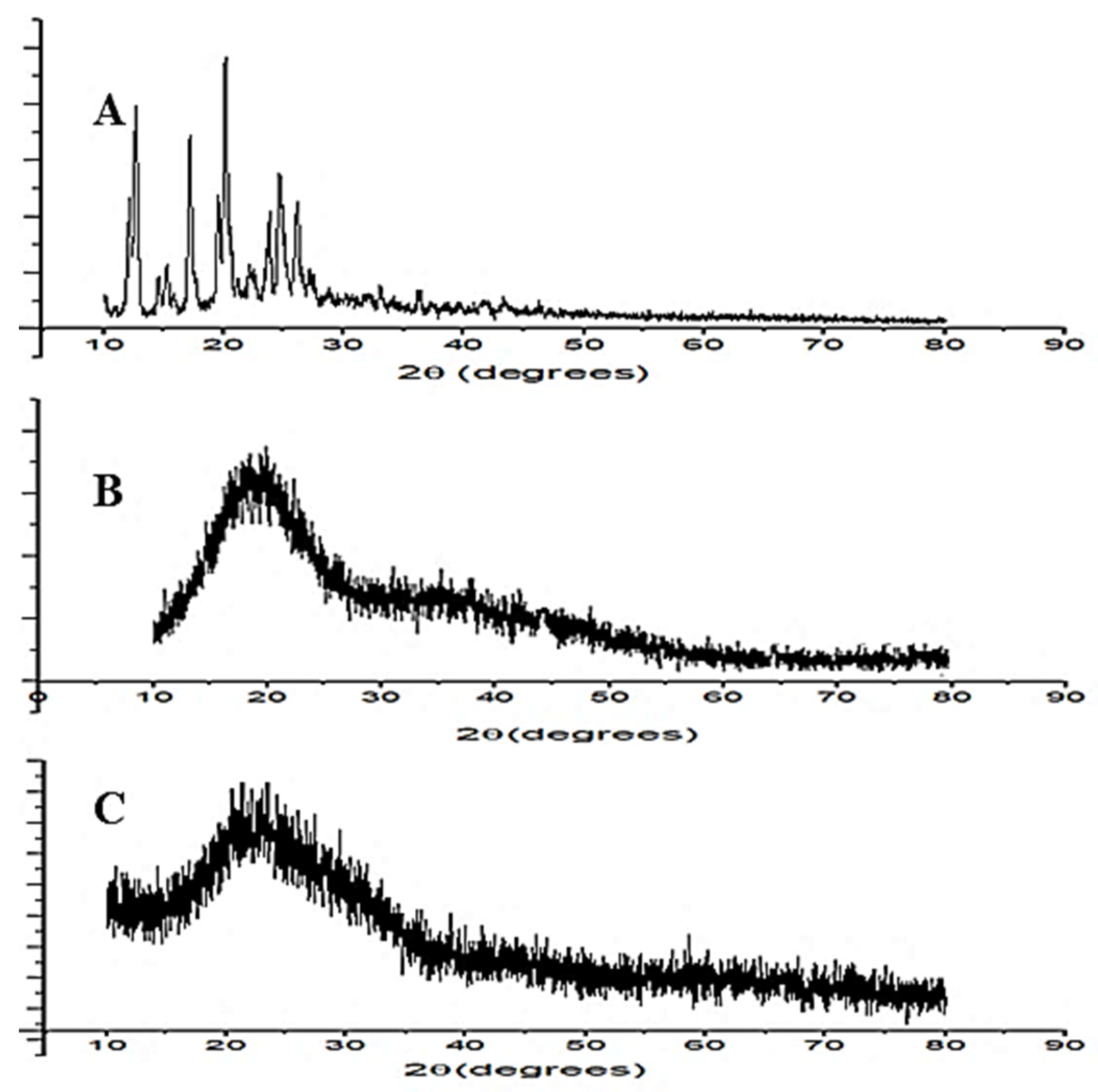
2.2.6. X-ray Diffraction Study (XRD)
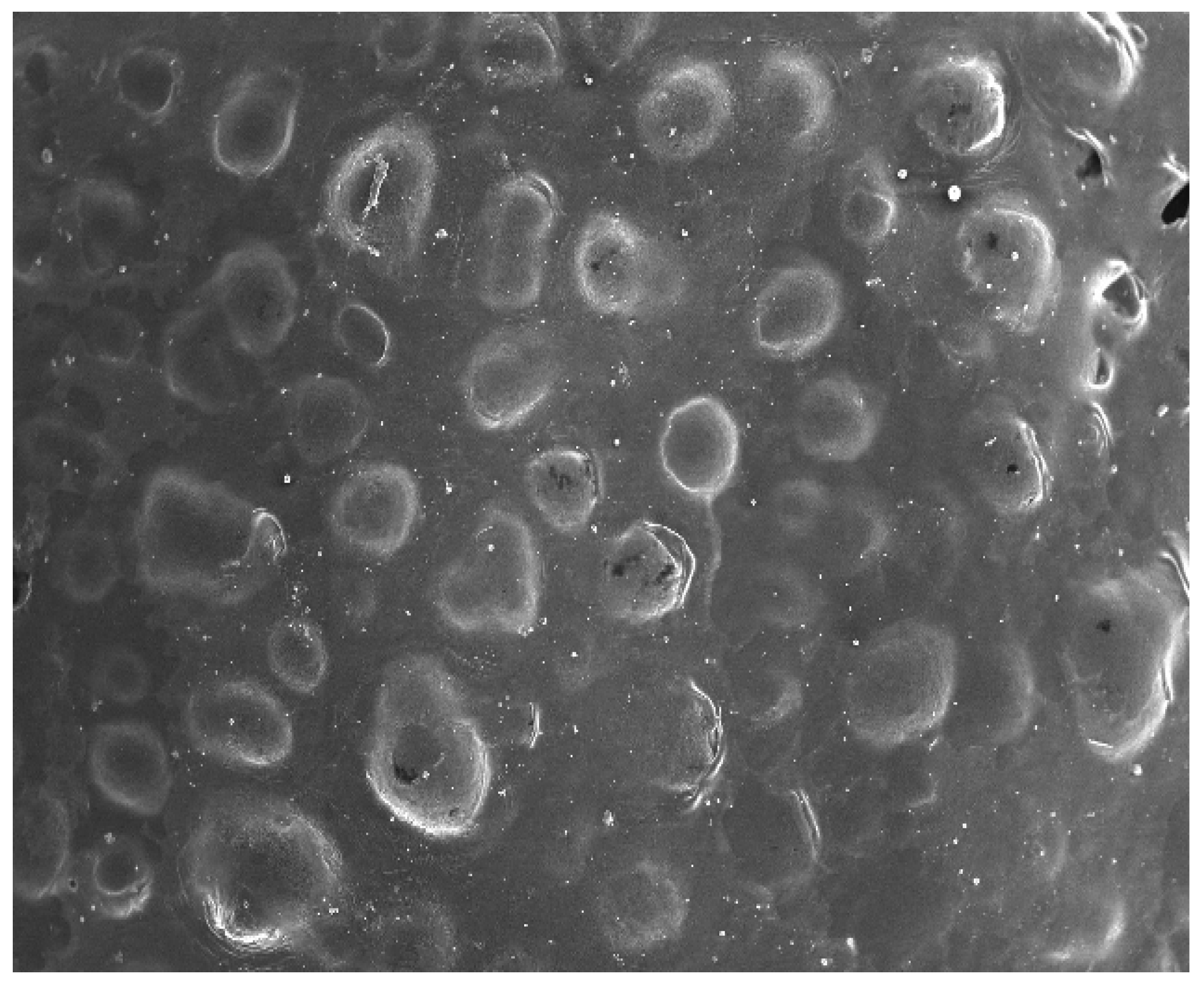
2.2.7. Surface Morphology
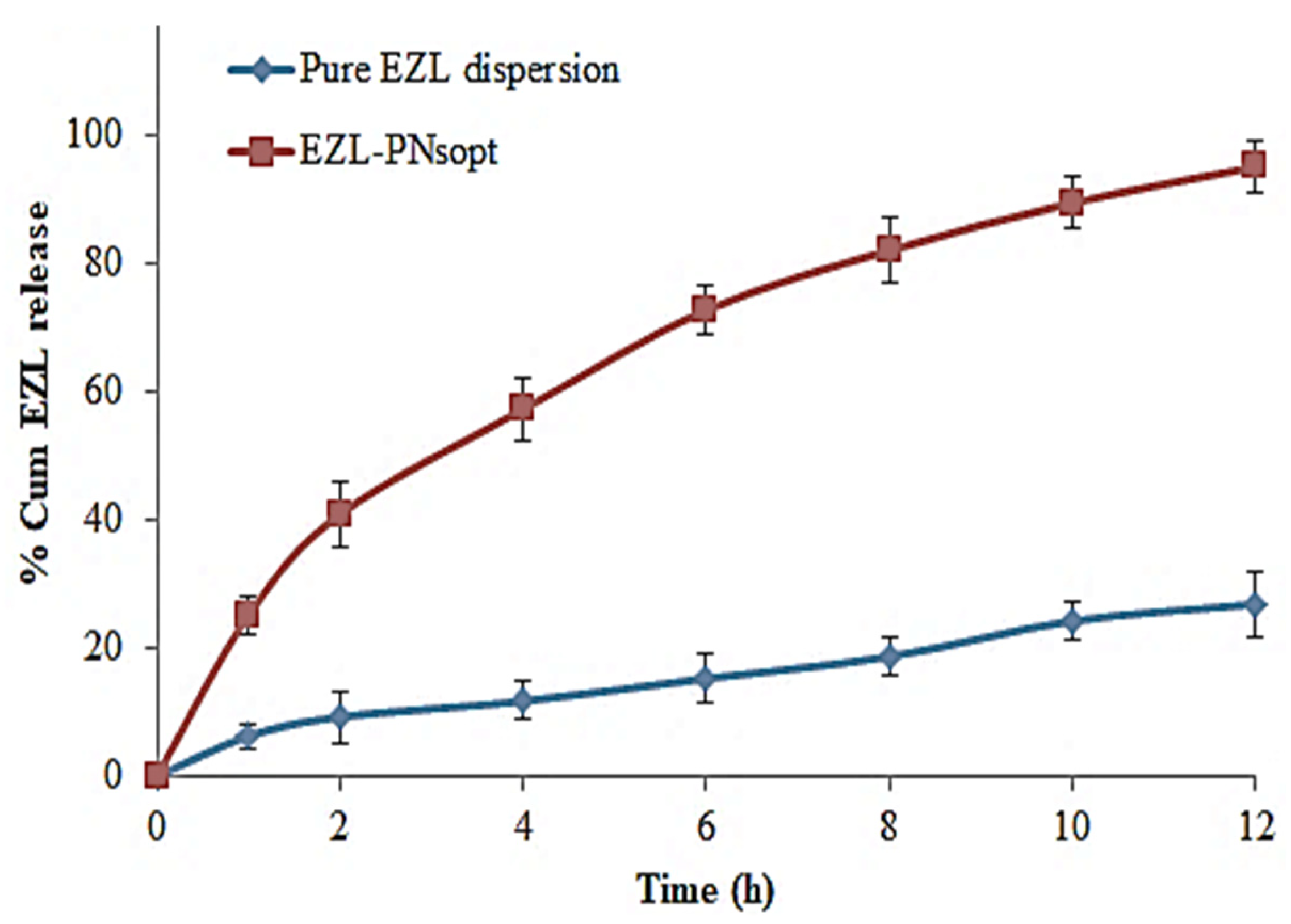
2.2.8. In-Vitro Release of EZL
2.2.9. Ex Vivo Permeability Study
2.2.10. In-Vivo Study
2.2.11. Pharmacokinetic Study
2.2.12. In Vivo Pharmacodynamic Study
2.2.13. Statistical Analysis
3. Result and Discussion
3.1. Optimization and Validation
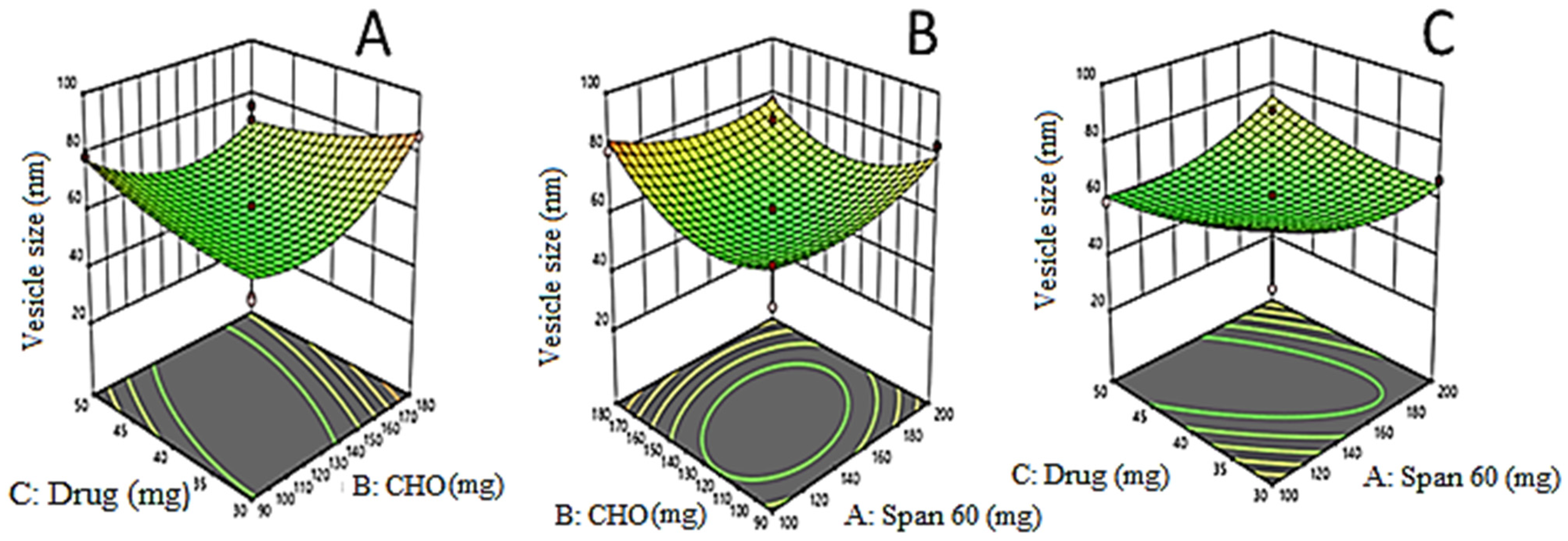
3.2. Effects of Formulation Variables on Vesicle Size (Y1)
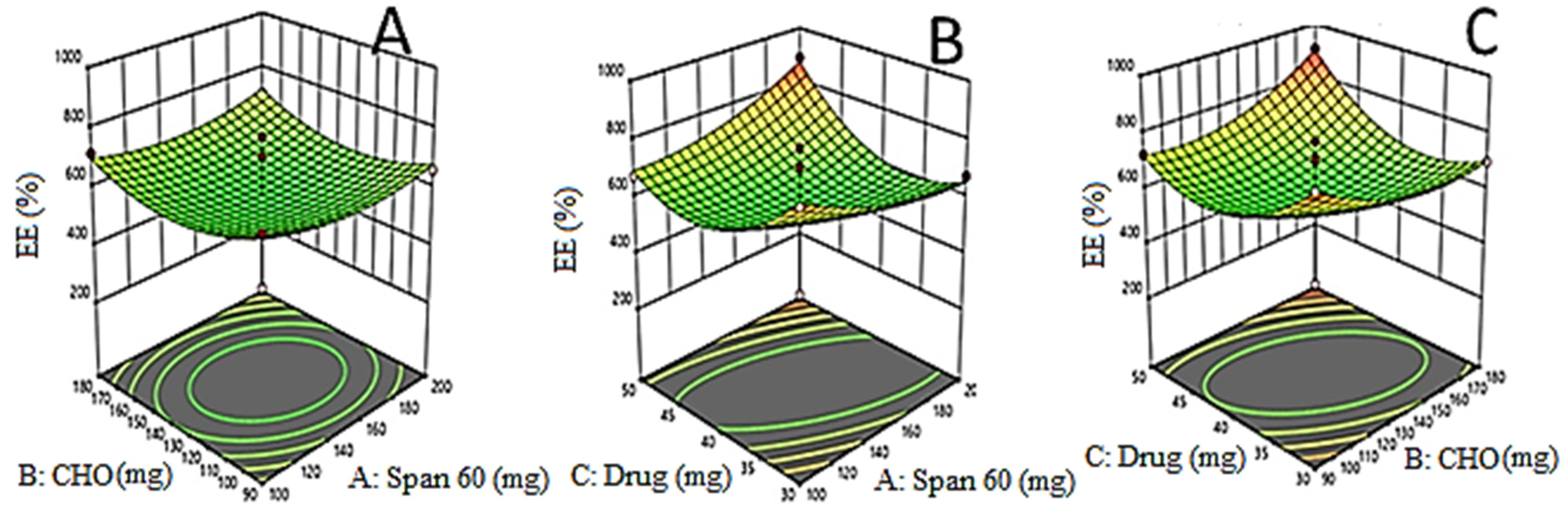
3.3. Effects of Formulation Variables on Drug Entrapment Efficiency (Y2)
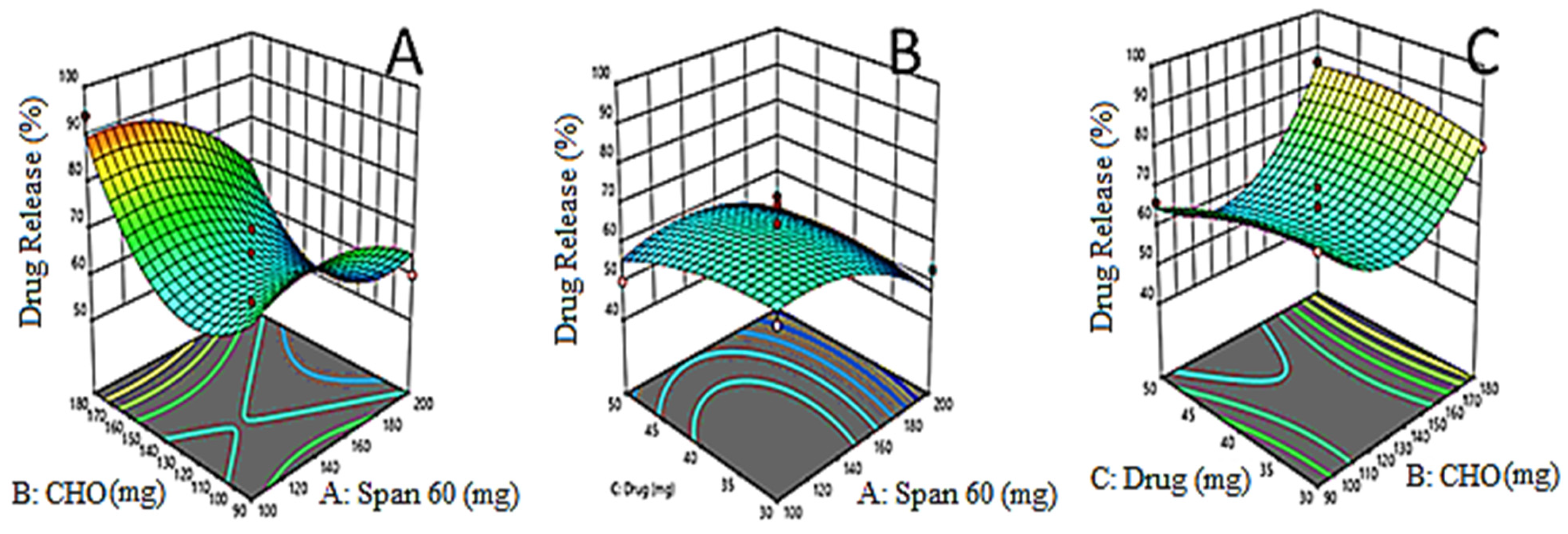
3.4. Effects of Formulation Variables on Drug Release (Y3)
3.5. Point Prediction Optimization
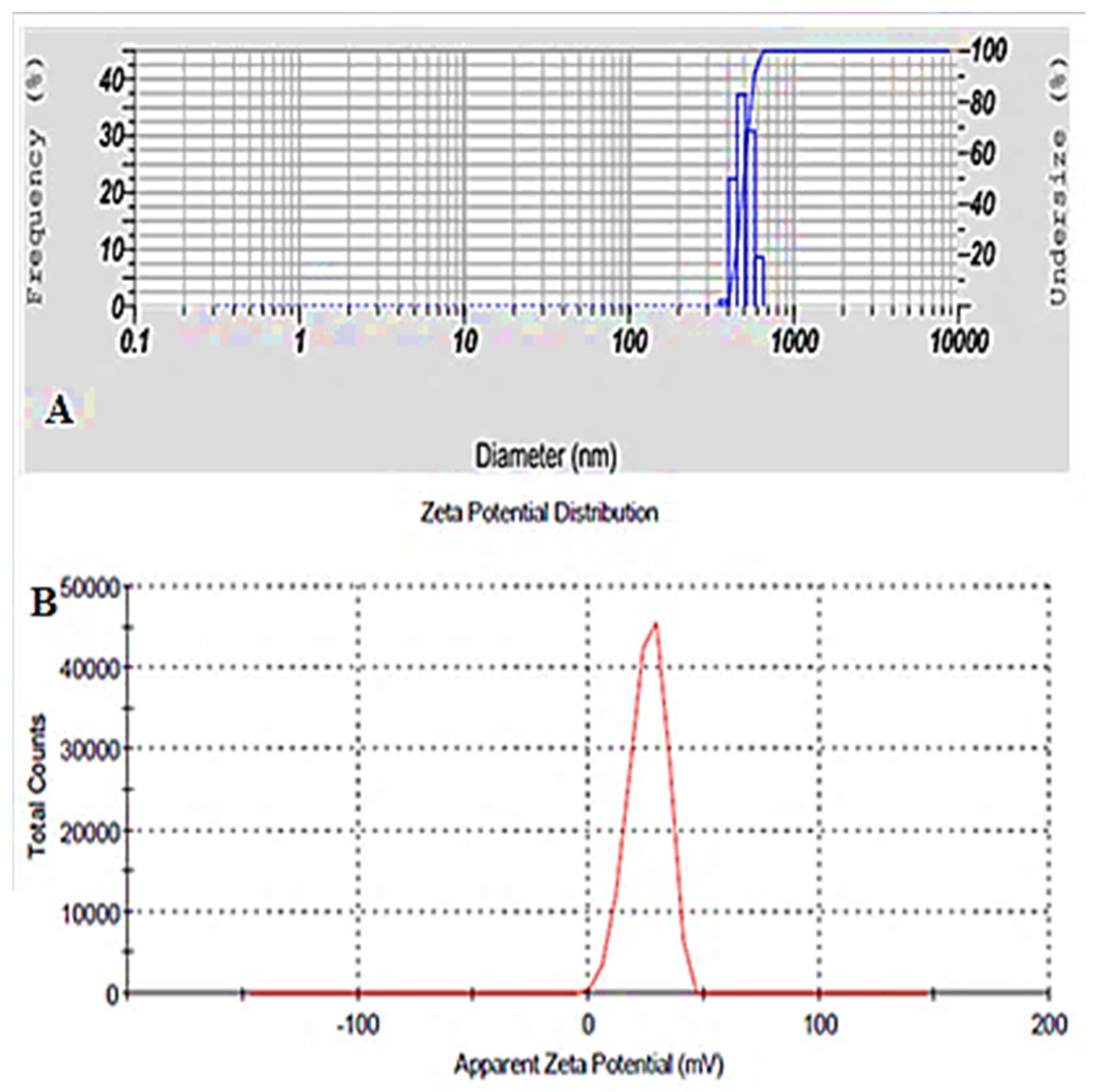
3.6. Vesicle Size, Zeta Potential and Surface Morphology
3.7. Entrapment Efficiency (EE %)
3.8. Thermal Transition Analysis
3.9. X-ray Diffraction Study
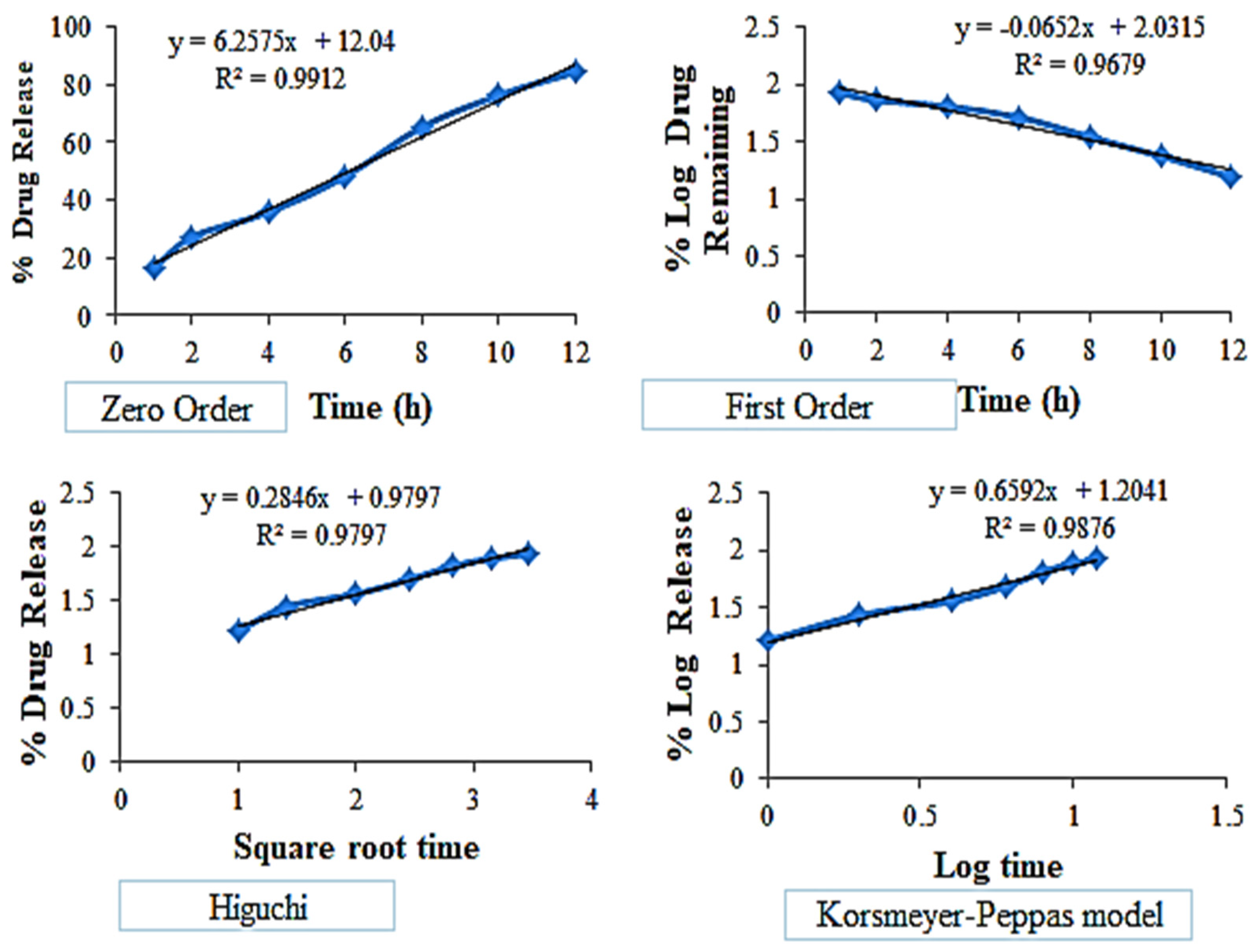
3.10. In Vitro Release of EZL
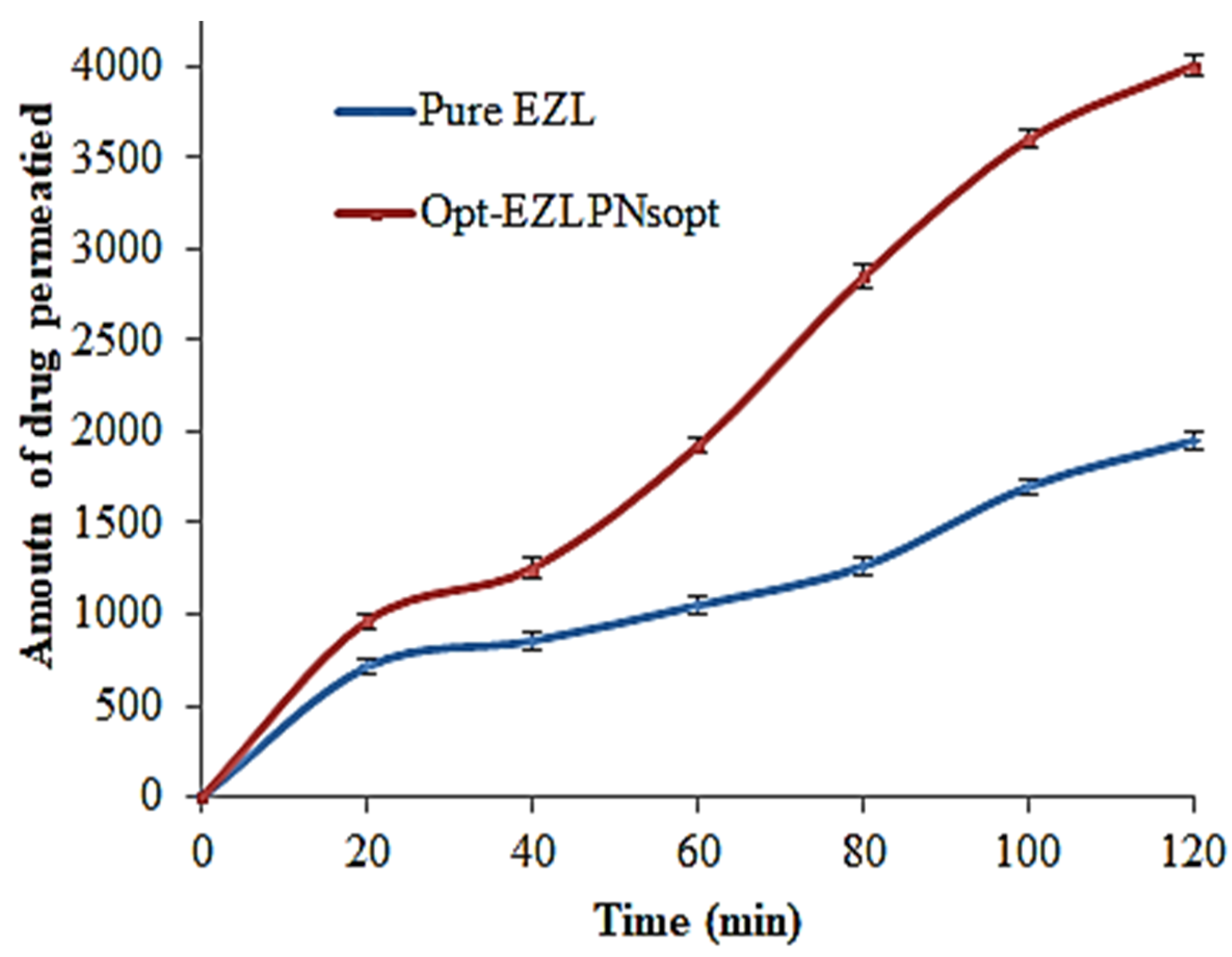
3.11. Ex Vivo Intestine Permeation Study
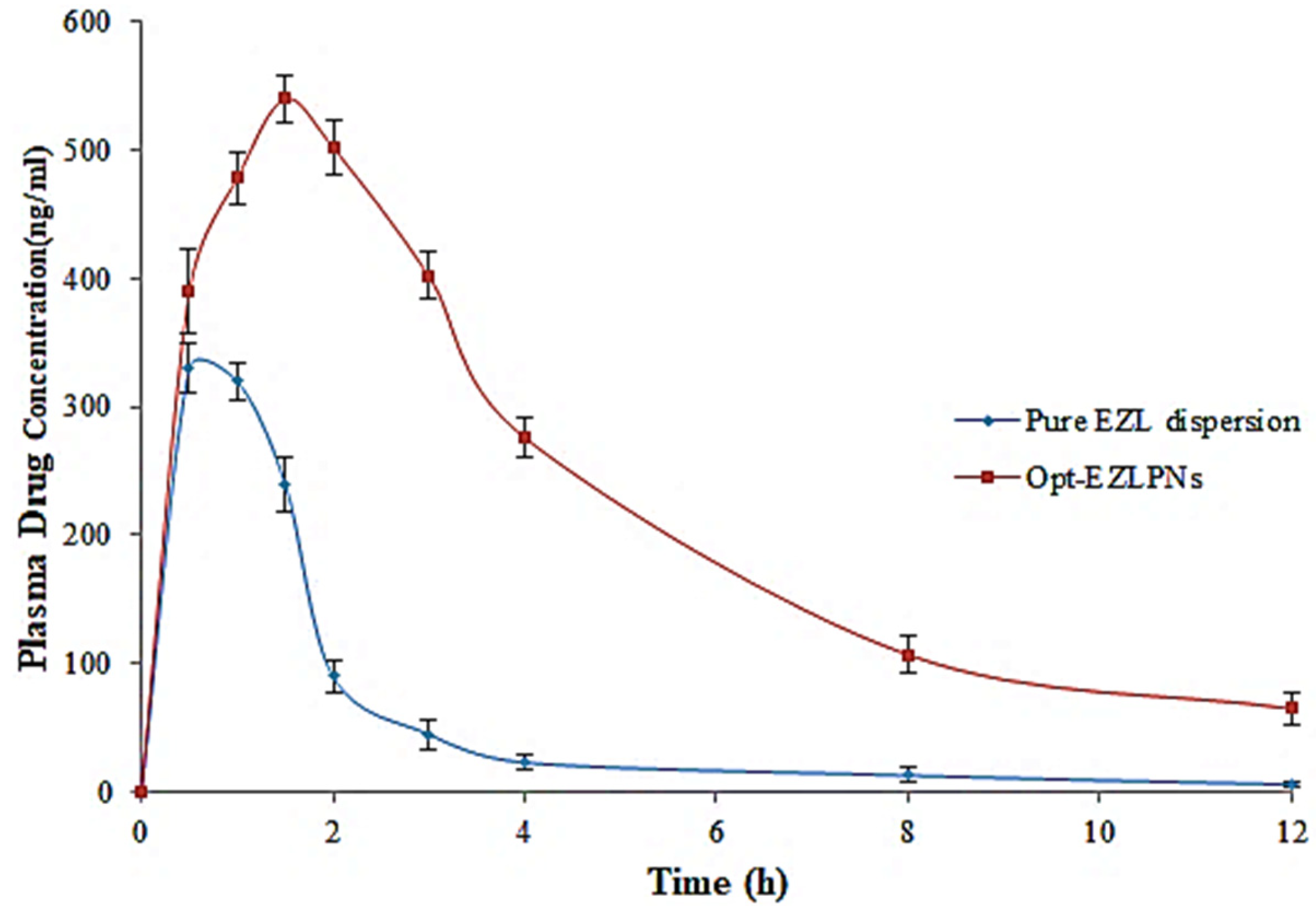
3.12. Pharmacokinetic Studies
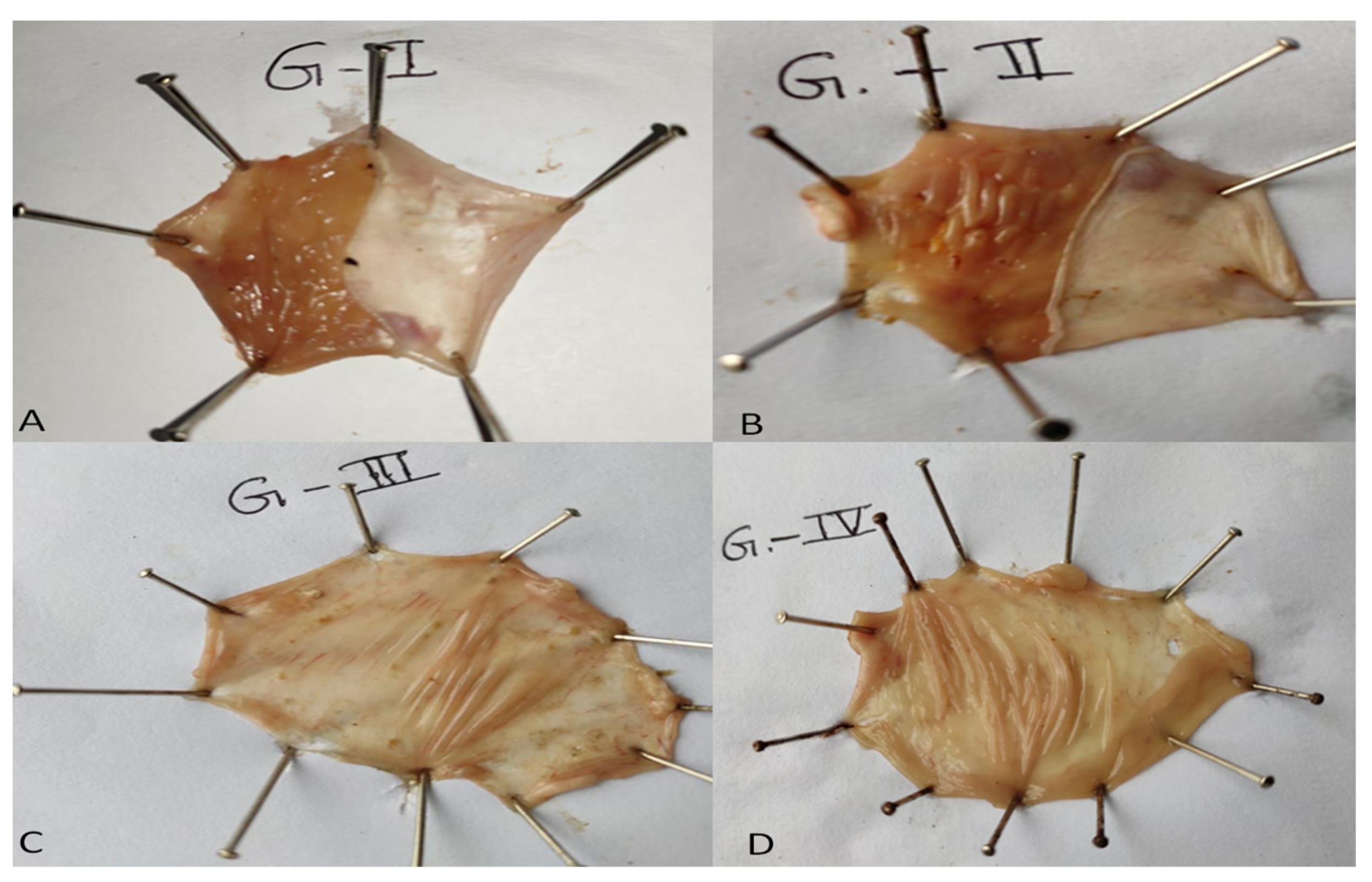
3.13. In Vivo Pharmacodynamic Study
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Goldstein, J.L.; Hochberg, M.C.; Fort, J.G.; Zhang, Y.; Hwang, C.; Sostek, M. Clinical trial: The incidence of NSAID-associated endoscopic gastric ulcers in patients treated with PN 400 (naproxen plus esomeprazole magnesium) vs. enteric-coated naproxen alone. Aliment. Pharmacol. Ther. 2010, 32, 401–413. [Google Scholar] [CrossRef]
- Kan, S.L.; Lu, J.; Liu, J.P.; Zhao, Y. Preparation and in vitro/in vivo evaluation of esomeprazole magnesium-modified release pellets. Drug Deliv. 2016, 23, 856–863. [Google Scholar] [CrossRef] [PubMed]
- Pantić, M.; Kravanja, K.A.; Knez, Ž.; Novak, Z. Influence of the Impregnation Technique on the Release of Esomeprazole from Various Bioaerogels. Polymers 2021, 13, 1882. [Google Scholar] [CrossRef] [PubMed]
- Omari, D.M. Formulation and In vitro/In vivo evaluation of esomeprazole enteric coated minitablets. J. Drug Deliv. Sci. Technol. 2017, 39, 156–165. [Google Scholar] [CrossRef]
- Barmpalexis, P.; Grypioti, A. Development of a new esomeprazole delayed release gastro-resistant pellet formulation with improved storage stability. Drug Dev. Ind. Pharm. 2018, 44, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.; Jung, J.; Yong, C.S.; Rhee, C.; Lee, M.; Han, J.; Park, K.; Kim, C. Formulation and in vivo evaluation of omeprazole buccal adhesive tablet. J. Control. Release 2000, 68, 405–412. [Google Scholar] [CrossRef]
- Kumar, P.; Ganure, A.L.; Subudhi, B.B.; Shukla, S. Preparation and characterization of pH-sensitive methyl methacrylate-gstarch/hydroxypropylated starch hydrogels: In vitro and in vivo study on release of esomeprazole magnesium. Drug Deliv. Transl. Res. 2015, 5, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Teja, M.N.R.; Pariyani, L.; Balamuralidhara, V.; Gupta, N.V. Formulation and Evaluation of Spray-Dried Esomeprazole Magnesium Microspheres. Trop. J. Pharm. Res. 2013, 12, 299–304. [Google Scholar] [CrossRef]
- Bhad, M.E.; Abdul, S.; Jaiswal, S.B.; Chandewar, A.V.; Jain, J.M.; Sakarkar, D.M. MUPS tablets–a brief review. Int. J. Pharm. Tech. Res. 2010, 2, 847–855. [Google Scholar]
- Lindberg, P.; Keeling, D.; Fryklund, J.; Andersson, T.; Lundborg, P.; Carlsson, E. Esomeprazole—Enhanced bio-availability, specificity for the proton pump and inhibition of acid secretion. Aliment. Pharmacol. Ther. 2003, 17, 481–488. [Google Scholar] [CrossRef]
- Mohanty, D.; Rani, M.J.; Haque, M.A.; Bakshi, V.; Jahangir, M.A.; Imam, S.S.; Gilani, S.J. Preparation and evaluation of transdermal naproxen niosomes: Formulation optimization to preclinical anti-inflammatory assessment on murine model. J. Liposome Res. 2020, 30, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Gilani, S.J.; Bin-Jumah, M.N.; Imam, S.S.; Alshehri, S.; Jahangir, M.A.; Zafar, A. Formulation and Optimization of Nano Lipid Based Oral Delivery Systems for Arthritis. Coatings 2021, 11, 548. [Google Scholar] [CrossRef]
- Jahangir, M.A.; Khan, R.; Imam, S.S. Formulation of sitagliptin-loaded oral polymeric nano scaffold: Process parameters evaluation and enhanced anti-diabetic performance. Artif. Cells Nanomed. Biotechnol. 2018, 46, 66–78. [Google Scholar] [CrossRef]
- Qumbar, M.; Imam, S.S.; Ali, J.; Ahmad, J.; Ali, A. Formulation and optimization of lacidipine loaded niosomal gel for transdermal delivery: In-vitro characterization and in-vivo activity. Biomed. Pharmacother. 2017, 93, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Nemr, A.A.; El-Mahrouk, G.M.; Badie, H.A. Development and evaluation of proniosomes to enhance the transdermal delivery of cilostazole and to ensure the safety of its application. Drug Dev. Ind. Pharm. 2021, 47, 403–415. [Google Scholar] [CrossRef]
- Rojanarat, W.; Nakpheng, T.; Thawithong, E.; Yanyium, N.; Srichana, T. Levofloxacin-proliposomes: Opportunities for use in lung tuberculosis. Pharmaceutics 2012, 4, 385–412. [Google Scholar] [CrossRef] [PubMed]
- Duangjit, S.; Opanasopit, P.; Rojanarata, T.; Ngawhirunpat, T. Evaluation of meloxicam-loaded cationic transfersomes as transdermal drug delivery carriers. AAPS PharmSciTech. 2013, 14, 133–140. [Google Scholar] [CrossRef]
- Semalty, A.; Semalty, M.; Rawat, B.S.; Singh, D.; Rawat, M.S. Development and evaluation of pharmacosomes of aceclofenac. Indian J. Pharm. Sci. 2010, 72, 576–581. [Google Scholar] [CrossRef]
- Sammour, R.M.; Taher, M.; Chatterjee, B.; Shahiwala, A.; Mahmood, S. Optimization of aceclofenac proniosomes by using different carriers, Part 1: Development and characterization. Pharmaceutics 2019, 11, 350. [Google Scholar] [CrossRef]
- Khatoon, M.; Shah, K.U.; Din, F.U.; Shah, S.U.; Rehman, A.U.; Dilawar, N.; Khan, A.N. Proniosomes derived niosomes: Recent advancements in drug delivery and targeting. Drug Deliv. 2017, 24, 56–69. [Google Scholar] [CrossRef]
- Radha, G.V.; Rani, T.S.; Sarvani, B. A review on proniosomal drug delivery system for targeted drug action. J. Basic Clin. Pharm. 2013, 4, 42. [Google Scholar] [CrossRef] [PubMed]
- Nowroozi, F.; Almasi, A.; Javidi, J.; Haeri, A.; Dadashzadeh, S. Effect of surfactant type, cholesterol content and various downsizing methods on the particle size of niosomes. Iran. J. Pharm. Res. IJPR 2018, 17, 1. [Google Scholar] [PubMed]
- Masjedi, M.; Montahaei, T. An illustrated review on nonionic surfactant vesicles (niosomes) as an approach in modern drug delivery: Fabrication, characterization, pharmaceutical, and cosmetic applications. J. Drug Deliv. Sci. Technol. 2021, 61, 102234. [Google Scholar] [CrossRef]
- Nasr, M. In Vitro and In Vivo Evaluation of Proniosomes Containing Celecoxib for Oral Administration. AAPS Pharmscitech 2010, 11, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Shehata, T.M.; Abdallah, M.H.; Ibrahim, M.M. Proniosomal Oral Tablets for Controlled Delivery and Enhanced Pharmacokinetic Properties of Acemetacin. AAPS Pharmscitech 2015, 16, 375–383. [Google Scholar] [CrossRef]
- Bomma, G.; Harika, S.M.; Babu, A.M.; Bakshi, V. Formulation Development and Evaluation of Proniosomal Powder of Candesartan. Anal. Chem. Lett. 2017, 7, 567–577. [Google Scholar] [CrossRef]
- Yuksel, N.; Bayindir, Z.S.; Aksakal, E.; Ozcelikay, A.T. In situ niosome forming maltodextrin proniosomes of candesartan cilexetil: In vitro and in vivo evaluations. Int. J. Biol. Macromol. 2016, 82, 453–463. [Google Scholar] [CrossRef]
- Ismail, S.; Khattab, A. Optimization of proniosomal itraconazole formulation using Box Behken design to enhance oral bioavailability. J. Drug Deliv. Sci. Technol. 2018, 45, 142–150. [Google Scholar] [CrossRef]
- Patil, M.; Pandit, P.; Udavant, P.; Sonawane, S.; Bhambere, D. Development and Optimization of Proniosomal Gel Containing Etodolac: In-vitro, Ex-vivo and In-vivo Evaluation. Ars Pharma. 2021, 62, 290–304. [Google Scholar] [CrossRef]
- Aboelwafa, A.A.; El-Setouhyand, D.A.; Nabil, A. Comparative study on the effects of some polyoxyethylene alkyl ether and sorbitan fatty acid ester surfactants on the performance of transdermal carvedilol proniosomal gel using experimental design. AAPS Pharmscitech 2010, 11, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.H.; Hou, J.P.; Liu, A.; Xie, Y.M.; Hu, X.; Song, X. HPLC Determination of Esomeprazole Magnesium in Tablets. Asian J. Chem. 2010, 22, 3711–3718. [Google Scholar]
- Sabiu, S.; Garuba, T.; Sunmonu, T.; Ajani, E.; Sulyman, A.; Nurain, I.; Balogun, A. Indomethacin-induced gastric ulceration in rats: Protective roles of Spondias mombin and Ficus exasperata. Toxicol. Rep. 2015, 2, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Thapa, S.K.; Lee, D.Y.; Chung, S.E.; Lim, J.Y.; Jeong, H.M.; Song, C.H.; Choi, Y.W.; Cho, S.M.; Nam, K.Y.; et al. Pharmacokinetics and Anti-Gastric Ulceration Activity of Oral Administration of Aceclofenac and Esomeprazole in Rats. Pharmaceutics 2018, 10, 152. [Google Scholar] [CrossRef] [PubMed]
- Hashim, I.I.A.; El-dahan, M.S.; Yusif, R.M.; Abd-ElGawad, A.E.G.H.; Arima, H. Potential use of niosomal hydrogel as an ocular delivery system for atenolol. Biol. Pharm. Bull. 2014, 37, 541–551. [Google Scholar] [CrossRef]
- Yoshioka, T.; Sternberg, B.; Florence, A.T. Preparation and properties of vesicles (niosomes) of sorbitan monoesters (Span 20, 40, 60 and 80) and a sorbitan triester (Span 85). Int. J. Pharm. 1994, 105, 1–6. [Google Scholar] [CrossRef]
- Ruwizhi, N.; Aderibigbe, B.A. The Efficacy of Cholesterol-Based Carriers in Drug Delivery. Molecules 2020, 25, 4330. [Google Scholar] [CrossRef] [PubMed]
- Lingan, M.A.; Sathali, A.A.H.; Kumar, M.R.V.; Gokila, A. Formulation and evaluation of topical drug delivery system containing clobetasol propionate niosomes. Sci. Revs. Chem. Commun. 2011, 1, 7–17. [Google Scholar]
- Mokhtar, M.; Sammour, O.A.; Hammad, M.A.; Megrab, N.A. Effect of some formulation parameters on flurbiprofen encapsulation and release rates of niosomes prepared from proniosomes. Int. J. Pharm. 2008, 361, 104–111. [Google Scholar] [CrossRef]
- Solanki, A.B.; Parikh, J.R.; Parikh, R.H. Formulation and optimization of piroxicam proniosomes by 3-factor, 3-level Box-Behnken design. AAPS Pharmscitech 2007, 8, E86. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Wei, M.; He, S.; Yuan, W.-E. Advances of Non-Ionic Surfactant Vesicles (Niosomes) and Their Application in Drug Delivery. Pharmaceutics 2019, 11, 55. [Google Scholar] [CrossRef] [PubMed]
- Shah, H.; Nair, A.B.; Shah, J.; Bharadia, P.; Al-Dhubiab, B.E. Proniosomal gel for transdermal delivery of lornoxicam: Optimization using factorial design and in vivo evaluation in rats. Daru 2019, 27, 59–70. [Google Scholar] [CrossRef]
- Gurrapu, A.; Jukanti, R.; Bobbala, S.R.; Kanuganti, S.; Jeevan, J.B. Improved oral delivery of valsartan from maltodextrin based proniosome powders. Adv. Powder Technol. 2012, 23, 583–590. [Google Scholar] [CrossRef]
- Nandy, B.C.; Mazumder, B. Formulation and characterizations of delayed release multi particulates system of indomethacin: Optimization by response surface methodology. Curr. Drug Deliv. 2014, 11, 72–86. [Google Scholar] [CrossRef] [PubMed]
- Bendas, E.R.; Abdelbary, A.A. Instantaneous enteric nano-encapsulation of omeprazole: Pharmaceutical and pharmacological evaluation. Int. J. Pharm. 2014, 468, 97–104. [Google Scholar] [CrossRef]
- Raffin, R.P.; Colomé, L.M.; Schapoval, E.E.S.; Jornada, D.S.; Pohlmann, A.R.; Guterres, S.S. Gastro-resistant microparticles containing sodium pantoprazole: Stability studies and in vivo anti-ulcer activity. Open Drug Deliv. J. 2007, 1, 28–35. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factors | Levels Used, Actual Coded | ||
|---|---|---|---|
| Independent Variables | Low (−1) | Mid (0) | High (+1) |
| X1 = Span 60 (mg) | 100 | 150 | 200 |
| X2 = Cholesterol (mg) | 90 | 135 | 180 |
| X3 = Drug amount (mg) | 30 | 40 | 50 |
| Dependent Variables | Goals | ||
| Y1 = Vesicle size (nm) | Minimize | ||
| Y2 = Entrapment efficiency (%) | Maximize | ||
| Y3 = Drug release (%) | Maximize | ||
| Formulation Code | Span 60 (X1) | Cholesterol (X2) | Drug (X3) | Vesicle Size (Y1, nm) | Entrapment Efficiency (Y2, %) | Drug Release (Y3, %) |
|---|---|---|---|---|---|---|
| EZL-PNs1 | 1 | 1 | 0 | 723 ± 12.32 | 78.57 ± 4.56 | 66 ± 2.1 |
| EZL-PNs2 | −1 | −1 | 0 | 905 ± 13.21 | 75.38 ± 3.13 | 86 ± 1.1 |
| EZL-PNs3 | 0 | 0 | 0 | 660 ± 12.16 | 82.73 ± 3.44 | 91 ± 2.4 |
| EZL-PNs4 | 0 | −1 | 1 | 698 ± 11.18 | 85.59 ± 4.68 | 80 ± 2.6 |
| EZL-PNs5 | 0 | 1 | 1 | 704 ± 11.11 | 76.93 ± 1.98 | 65 ± 1.1 |
| EZL-PNs6 | 1 | 0 | 1 | 771 ± 13.71 | 61.62 ± 1.98 | 70 ± 1.2 |
| EZL-PNs7 | 1 | −1 | 0 | 848 ± 12.40 | 59.37 ± 1.09 | 75 ± 2.4 |
| EZL-PNs8 | 1 | 0 | −1 | 889 ± 12.10 | 70.82 ± 3.38 | 53 ± 2.3 |
| EZL-PNs9 | 0 | 0 | 0 | 667 ± 10.81 | 81.65 ± 2.35 | 93 ± 2.1 |
| EZLPNs10 | −1 | 0 | −1 | 720 ± 10.91 | 70.32 ± 2.86 | 73 ± 2.2 |
| EZL-PNs11 | −1 | 0 | 1 | 674 ± 12.92 | 66.82 ± 0.42 | 53 ± 2.4 |
| EZLPNs12 | −1 | 1 | 0 | 250 ± 12.36 | 27.92 ± 2.06 | 56 ± 2.3 |
| EZL-PNs13 | 0 | 1 | −1 | 715 ± 13.21 | 80.85 ± 2.05 | 80 ± 2.2 |
| EZL-PNs14 | 0 | 0 | 0 | 663 ± 11.09 | 81.23 ± 0.63 | 92 ± 2.7 |
| EZL-PNs15 | 0 | −1 | −1 | 825 ± 12.31 | 80.85 ± 2.05 | 62 ± 2.8 |
| Source | Vesicle Size | Entrapment Efficiency | Drug Release |
|---|---|---|---|
| Model | Quadratic | Quadratic | Quadratic |
| Adjusted R2 | 0.9920 | 0.9521 | 0.9819 |
| R2 | 0.9934 | 0.9656 | 0.9865 |
| Predicted R2 | 0.9813 | 0.9543 | 0.9802 |
| % CV | 3.03 | 3.87 | 5.65 |
| Adequate precision | 65.05 | 28.43 | 43.87 |
| Standard Deviation | 6.87 | 3.14 | 3.23 |
| Pharmacokinetic Parameter | Pure EZL Dispersion | Opt-EZLPNs |
|---|---|---|
| t1/2 (h) | 1.35 ± 0.12 | 3.27 ± 0.16 |
| Cmax (ng) | 330 ± 20 | 540 ± 18 |
| Tmax (h) | 0.5 ± 0.03 | 1.5 ± 0.04 |
| AUC0-t (ng. h/mL) | 679 ± 12.05 | 2730.5 ± 8.45 |
| Ke (h−1) | 0.351 | 0.211 |
| AUC0-∞ (ng. h/mL) | 696.08 ± 21 | 3038.03 ± 19 |
| AUMC0-t (µg. h2/mL) | 1482.5 ± 23 | 10,444.5 ± 24 |
| AUMC0-∞ (µg. h2/mL) | 1736.11 ± 25 | 15,589.91 ± 19 |
| Group | Ulcer Index % | Ulcer Protection % |
|---|---|---|
| Control (Group-I) | 1.02 ± 0.1 | - |
| Indomethacin Induced (Group-II) | 7.85 ± 0.13 | - |
| EZL-Standard (Group-III) | 4.41 ± 0.3 | 43.82 |
| EZL-PNopt (Group-IV) | 3.01 ± 0.12 | 61.65 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohanty, D.; Zafar, A.; Jafar, M.; Upadhyay, A.K.; Haque, M.A.; Gupta, J.K.; Bakshi, V.; Ghoneim, M.M.; Alshehri, S.; Jahangir, M.A.; et al. Development, In-Vitro Characterization and Preclinical Evaluation of Esomeprazole-Encapsulated Proniosomal Formulation for the Enhancement of Anti-Ulcer Activity. Molecules 2022, 27, 2748. https://doi.org/10.3390/molecules27092748
Mohanty D, Zafar A, Jafar M, Upadhyay AK, Haque MA, Gupta JK, Bakshi V, Ghoneim MM, Alshehri S, Jahangir MA, et al. Development, In-Vitro Characterization and Preclinical Evaluation of Esomeprazole-Encapsulated Proniosomal Formulation for the Enhancement of Anti-Ulcer Activity. Molecules. 2022; 27(9):2748. https://doi.org/10.3390/molecules27092748
Chicago/Turabian StyleMohanty, Dibyalochan, Ameeduzzafar Zafar, Mohammed Jafar, Atul Kumar Upadhyay, Mohammad Akiful Haque, Jeetendra Kumar Gupta, Vasudha Bakshi, Mohammed M. Ghoneim, Sultan Alshehri, Mohammed Asadullah Jahangir, and et al. 2022. "Development, In-Vitro Characterization and Preclinical Evaluation of Esomeprazole-Encapsulated Proniosomal Formulation for the Enhancement of Anti-Ulcer Activity" Molecules 27, no. 9: 2748. https://doi.org/10.3390/molecules27092748
APA StyleMohanty, D., Zafar, A., Jafar, M., Upadhyay, A. K., Haque, M. A., Gupta, J. K., Bakshi, V., Ghoneim, M. M., Alshehri, S., Jahangir, M. A., & Ansari, M. J. (2022). Development, In-Vitro Characterization and Preclinical Evaluation of Esomeprazole-Encapsulated Proniosomal Formulation for the Enhancement of Anti-Ulcer Activity. Molecules, 27(9), 2748. https://doi.org/10.3390/molecules27092748