
One-Pot Synthesis of (E)-2-(3-Oxoindolin-2-ylidene)-2-arylacetonitriles

 , , ,
, , ,
Abstract
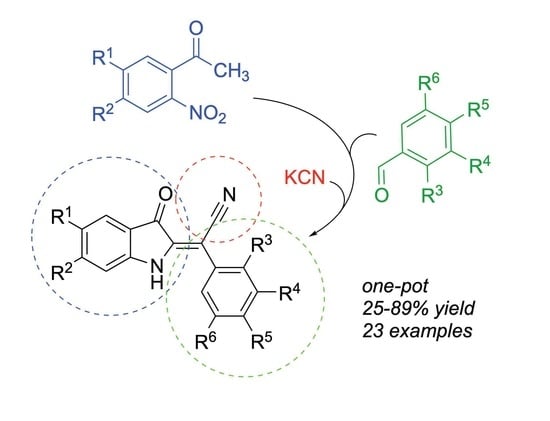
:
1. Introduction
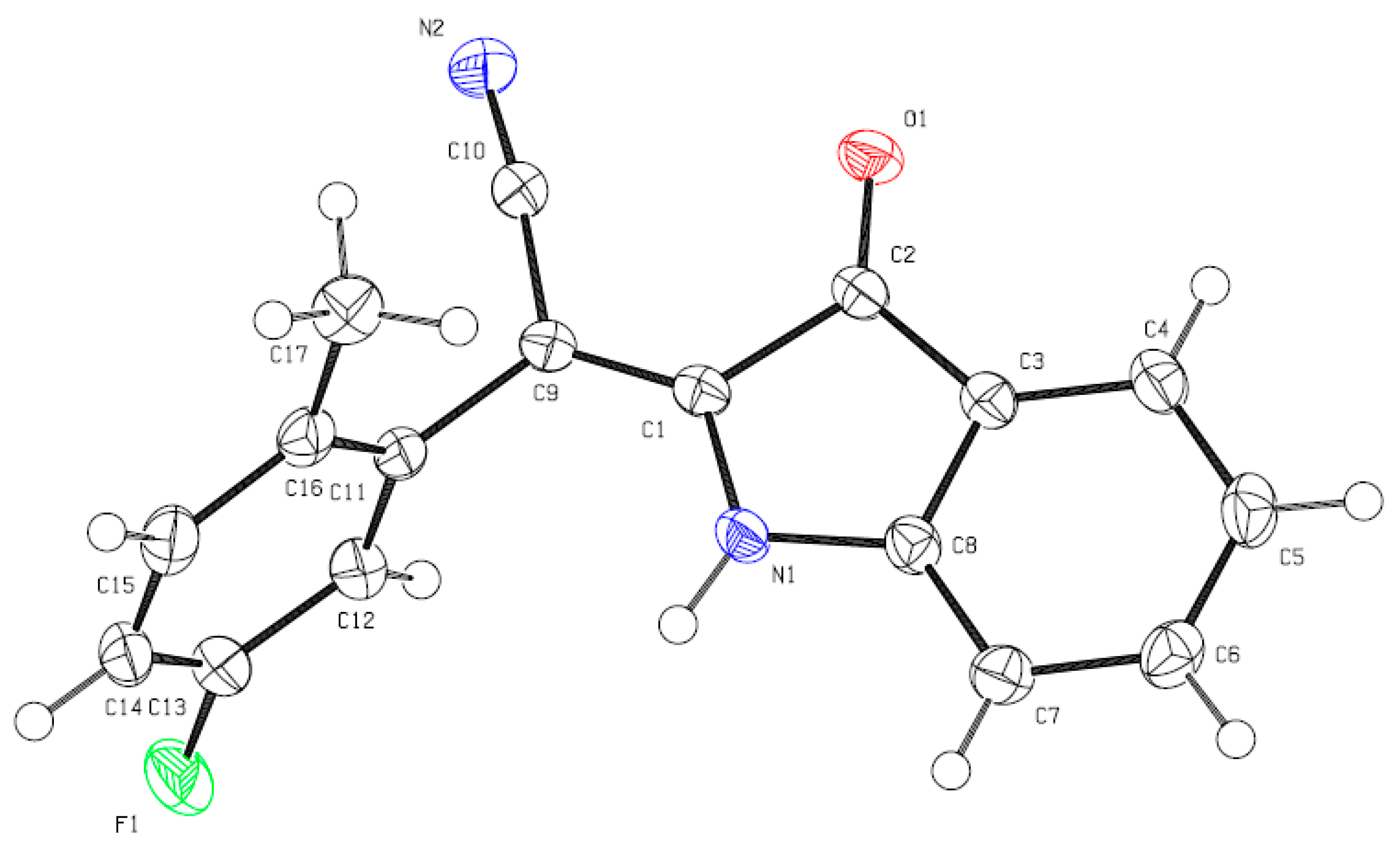
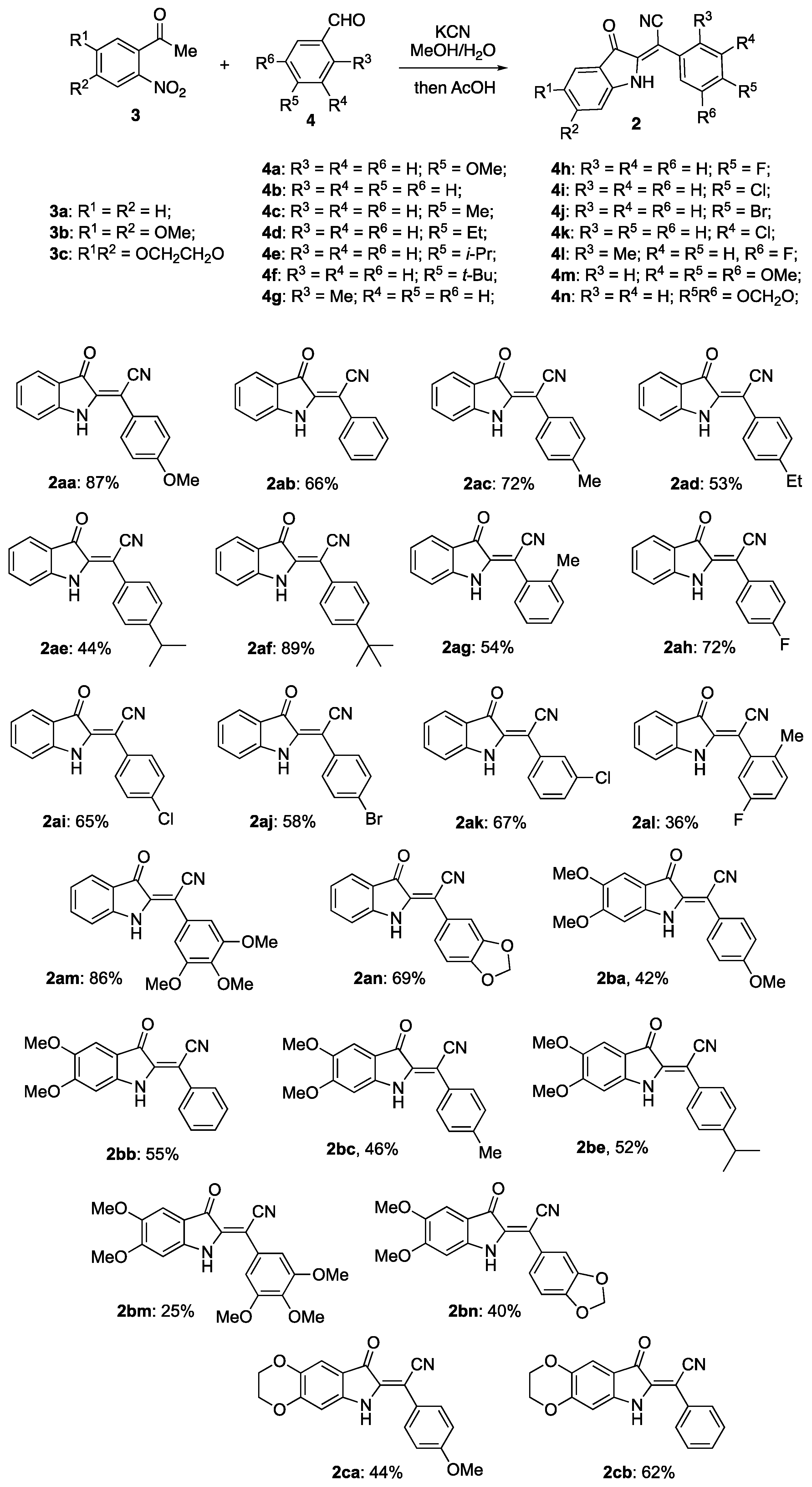
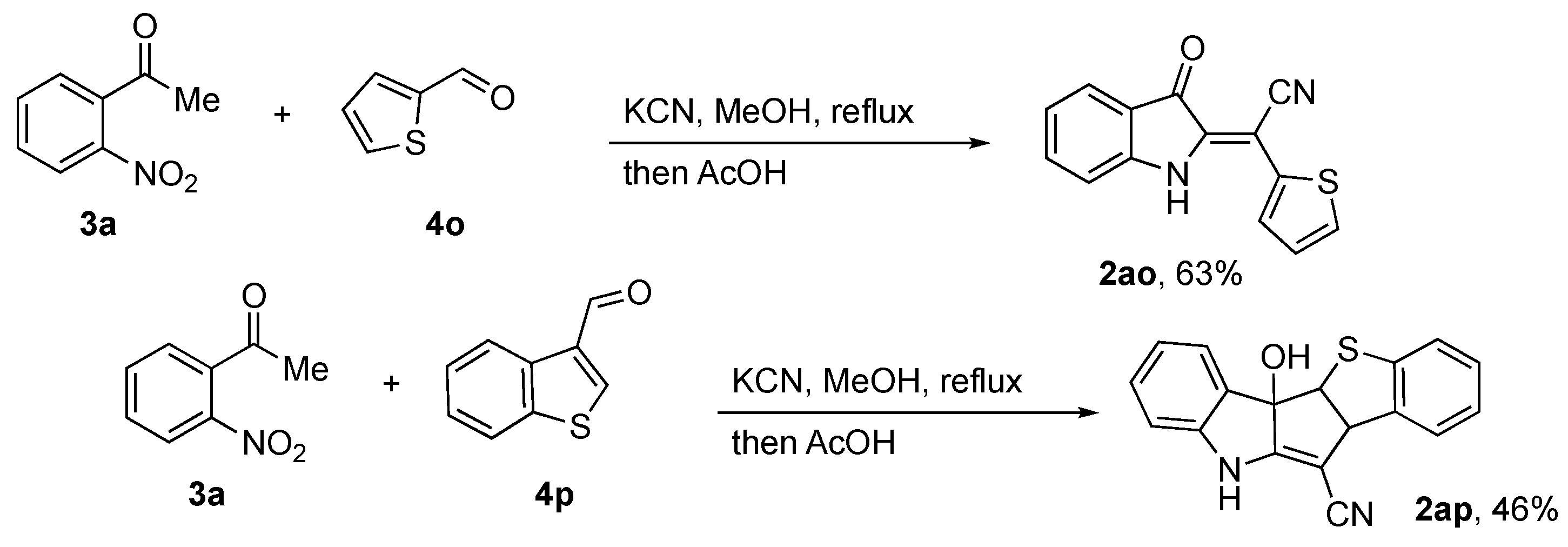
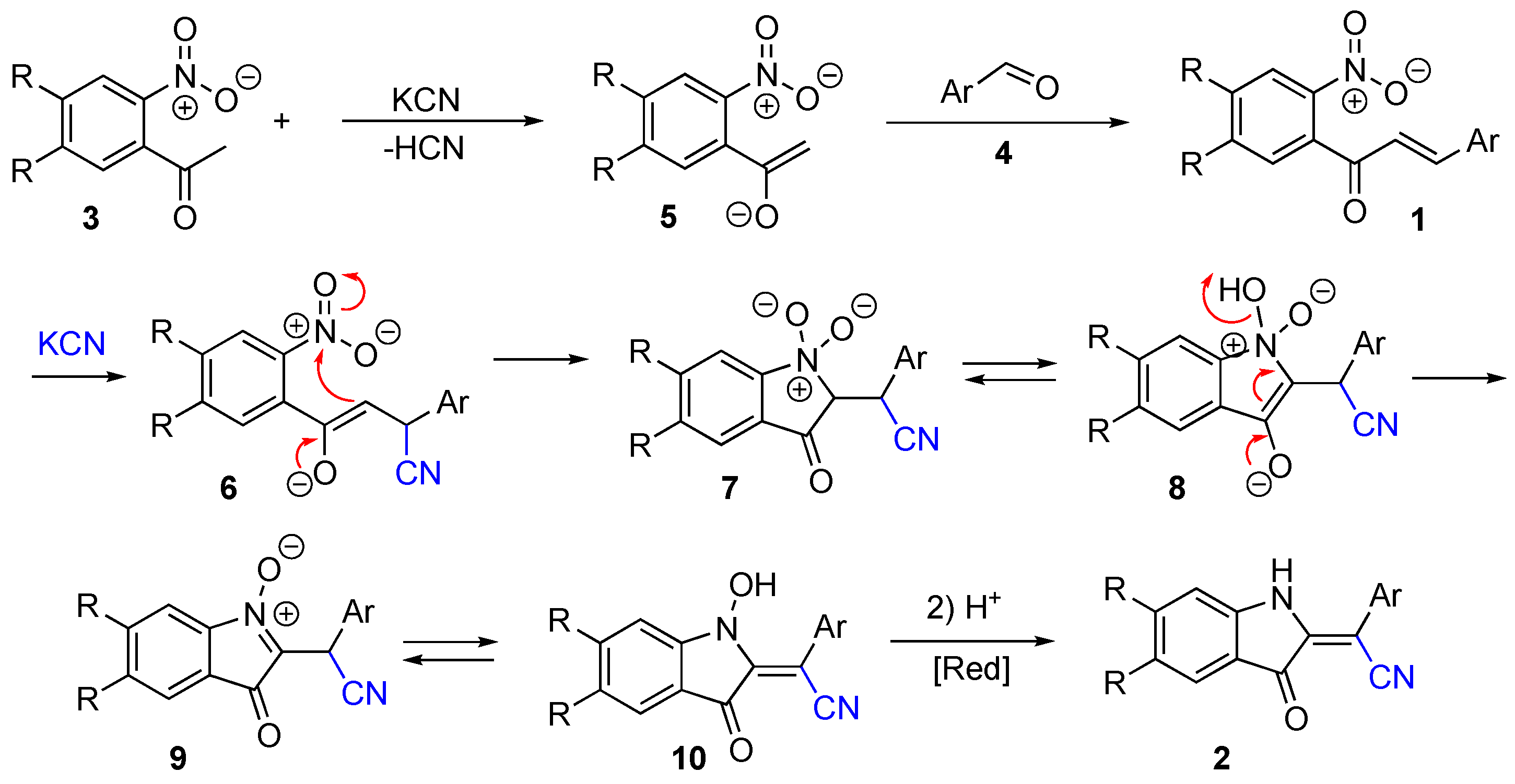
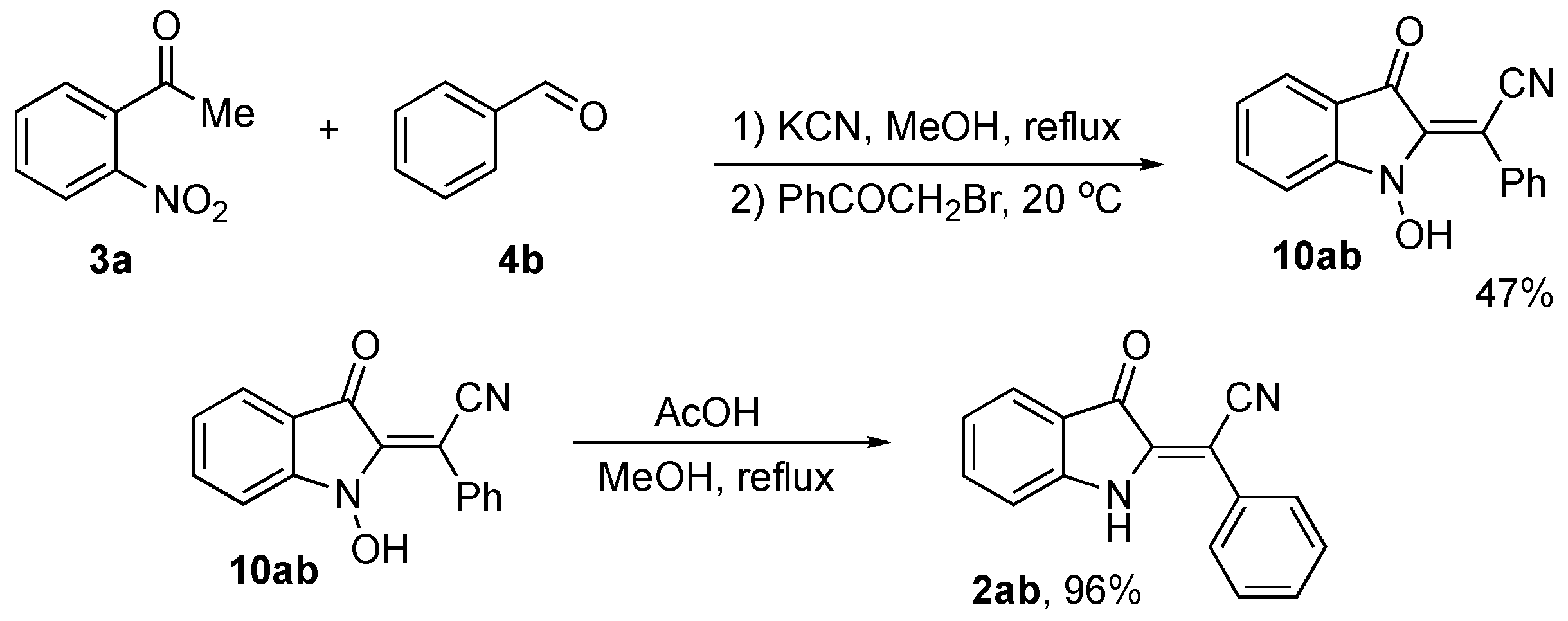
2. Results and Discussion
3. Conclusions
4. Experimental Part
General
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Hoessel, R.; Leclerc, S.; Endicott, J.A.; Nobel, M.E.M.; Lawrie, A.; Tunnah, P.; Leost, M.; Damiens, E.; Marie, D.; Marko, D.; et al. Indirubin, the active constituent of a Chinese antileukemia medicine, inhibits cyclin-dependent kinases. Nat. Cell Biol. 1999, 1, 60–67. [Google Scholar] [CrossRef]
- Leclerc, S.; Garnier, M.; Hoessel, R.; Marko, D.; Bibb, J.A.; Snyder, G.L.; Greengard, P.; Biernat, J.; Wu, Y.-Z.; Mandelkow, E.-M.; et al. Indirubins inhibit glycogen synthase kinase-3β and CDK5/P25, two protein kinases involved in abnormal tau phosphorylation in Alzheimer’s disease. A property common to most cyclin-dependent kinase inhibitors? J. Biol. Chem. 2001, 276, 251–260. [Google Scholar] [CrossRef] [Green Version]
- Meijer, L.; Skaltsounis, A.-L.; Magiatis, P.; Polychronopoulos, P.; Knockaert, M.; Leost, M.; Ryan, X.P.; Vonica, C.A.; Brivanlou, A.; Dajani, R.; et al. GSK-3-Selective Inhibitors Derived from Tyrian Purple Indirubins. Chem. Biol. 2003, 10, 1255–1266. [Google Scholar] [CrossRef] [Green Version]
- Polychronopoulos, P.; Magiatis, P.; Skaltsounis, A.-L.; Myrianthopoulos, V.; Mikros, E.; Tarricone, A.; Musacchio, A.; Roe, S.M.; Pearl, L.; Leost, M.; et al. Structural Basis for the Synthesis of Indirubins as Potent and Selective Inhibitors of Glycogen Synthase Kinase-3 and Cyclin-Dependent Kinases. J. Med. Chem. 2004, 47, 935–946. [Google Scholar] [CrossRef]
- Seidler, J.; McGovern, S.L.; Doman, T.N.; Shoichet, B.K. Identification and prediction of promiscuous aggregating inhibitors among known drugs. J. Med. Chem. 2003, 46, 4477–4486. [Google Scholar] [CrossRef]
- Wang, L.; Zhou, G.-B.; Liu, P.; Song, J.-H.; Liang, Y.; Yan, X.-J.; Xu, F.; Wang, B.-S.; Mao, J.-H.; Shen, Z.-X.; et al. Dissection of mechanisms of Chinese medicinal formula Realgar-Indigo naturalis as an effective treatment for promyelocytic leukemia. Proc. Natl. Acad. Sci. USA 2008, 105, 4826–4831. [Google Scholar] [CrossRef] [Green Version]
- Gein, V.L.; Tatarinov, V.V.; Rassudikhina, N.A.; Vakhrin, M.I.; Voronina, E.V. Synthesis and antimicrobial activity of 2-aroylmethylene-6-hydroxy-2,3-dihydroindol-3-ones. Pharm. Chem. J. 2011, 45, 231–232. [Google Scholar] [CrossRef]
- Lack, N.A.; Axerio-Cilies, P.; Tavassoli, P.; Han, F.G.; Chan, K.H.; Feau, C.; LeBlanc, E.; Guns, E.T.; Guy, R.K.; Rennie, P.S.; et al. Targeting the Binding Function 3 (BF3) Site of the Human Androgen Receptor through Virtual Screening. J. Med. Chem. 2011, 54, 8563–8573. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.-Q.; Mao, S.-C.; Zhang, H.-Y.; Yu, X.-Q.; Feng, M.-T.; Wang, B.; Feng, L.-H.; Guo, Y.-W. Racemosins A and B, two novel bisindole alkaloids from the green alga Caulerpa racemosa. Fitoterapia 2013, 91, 15–20. [Google Scholar] [CrossRef]
- Medvedev, A.E.; Ivanov, A.S.; Kamyshanskaya, N.S.; Kirkel, A.Z.; Moskvitina, T.A.; Gorkin, V.Z.; Li, N.Y.; Marshakov, V.Y. Interaction of indole derivatives with monoamine oxidase A and B. Studies on the structure-inhibitory activity relationship. Biochem. Mol. Biol. Int. 1995, 36, 113–122. [Google Scholar]
- Medvedev, A.E.; Ivanov, A.S.; Veselovsky, A.V.; Skvortsov, V.S.; Archakov, A.I. QSAR Analysis of Indole Analogs as Monoamine Oxidase Inhibitors. J. Chem. Inf. Comput. Sci. 1996, 36, 664–671. [Google Scholar] [CrossRef]
- Ornano, L.; Donno, Y.; Sanna, C.; Ballero, M.; Serafini, M.; Bianco, A. Phytochemical study of Caulerpa racemosa (Forsk.) J. Agarth, an invading alga in the habitat of La Maddalena Archipelago. Nat. Prod. Res. 2014, 28, 1795–1799. [Google Scholar] [CrossRef]
- Roaiah, H.M.; Ahmed, K.M.; Fawzy, N.M.; Wietrzyk, J.; Pawlik, A.; Ali, M.M.; Soliman, A.M. Synthesis of novel acetamide derivatives and evaluation of their antiproliferative potency against different cancer cell lines. Int. J. Pharm. Sci. Rev. Res. 2016, 36, 129–136. [Google Scholar]
- Guo, C.; Schedler, M.; Daniliuc, C.G.; Glorius, F. N-Heterocyclic carbene-catalyzed formal [3+2] annulation reaction of enals. An efficient enantioselective access to spiro-heterocycles. Angew. Chem. Int. Ed. 2014, 53, 10232–10236. [Google Scholar] [CrossRef]
- Merour, J.-Y.; Chichereau, L.; Desarbre, E.; Gadonneix, P. Synthesis and reactivity of (3-oxo-2,3-dihydro-1H-indol-2-ylidene)acetic acid alkyl esters in Diels-Alder Reactions. Synthesis 1996, 519–524. [Google Scholar] [CrossRef]
- Sim, H.M.; Loh, K.Y.; Yeo, W.K.; Lee, C.Y.; Go, M.L. Aurones as Modulators of ABCG2 and ABCB1: Synthesis and Structure-Activity Relationships. ChemMedChem 2011, 6, 713–724. [Google Scholar] [CrossRef]
- Souard, F.; Okombi, S.; Beney, C.; Chevalley, S.; Valentin, A.; Boumendjel, A. 1-Azaaurones derived from the naturally occurring aurones as potential antimalarial drugs. Bioorg. Med. Chem. 2010, 18, 5724–5731. [Google Scholar] [CrossRef]
- An, Z.W.; Catellani, M.; Chiusoli, G.P. A new palladium-catalyzed synthesis of indoxyl derivatives. J. Organomet. Chem. 1990, 397, C31–C32. [Google Scholar] [CrossRef]
- Genelot, M.; Bendjeriou, A.; Dufaud, V.; Djakovitch, L. Optimized procedures for the one-pot selective syntheses of indoxyls and 4-quinolones by a carbonylative Sonogashira/cyclization sequence. Appl. Catal. A 2009, 369, 125–132. [Google Scholar] [CrossRef]
- Genelot, M.; Dufaud, V.; Djakovitch, L. Heterogeneous metallo-organocatalysis for the selective one-pot synthesis of 2-benzylidene-indoxyl and 2-phenyl-4-quinolone. Tetrahedron 2011, 67, 976–981. [Google Scholar] [CrossRef]
- Li, R.; Qi, X.; Wu, X.-F. A general and convenient palladium-catalyzed synthesis of benzylideneindolin-3-ones with formic acid as the CO source. Org. Biomol. Chem. 2017, 15, 6905–6908. [Google Scholar] [CrossRef] [PubMed]
- Gein, V.L.; Demeneva, A.V.; Rassudikhina, N.A.; Vakhrin, M.I. Synthesis of 2-aroylmethylidene-6-hydroxy-2,3-dihydroindol-3-ones. Russ. J. Org. Chem. 2006, 42, 617–618. [Google Scholar] [CrossRef]
- Aksenov, N.A.; Aksenov, D.A.; Arutiunov, N.A.; Aksenova, D.S.; Aksenov, A.V.; Rubin, M. Unexpected cyclization of ortho-nitrochalcones into 2-alkylideneindolin-3-ones. RSC Adv. 2020, 10, 18440–18450. [Google Scholar] [CrossRef]
- Migita, M.; Okada, T.; Mataga, N.; Sakata, Y.; Misumi, S.; Nakashima, N.; Yoshihara, K. Picosecond laser spectroscopy of intramolecular heteroexcimer systems. Time-resolved fluorescence studies of p-(CH3)2NC6H4-(CH2)n-(9-anthryl), p-(CH3)2NC6H4-(CH2)n-(1-pyrenyl) systems and 9,9’-bianthryl. Bull. Chem. Soc. Jpn. 1981, 54, 3304–3311. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Hu, Z.; Zhang, X.; Dong, J.; Liu, J.-B.; Chen, D.-Z.; Xu, X. Tandem Synthesis of Pyrrolo[2,3-b]quinolones via Cadogen-Type Reaction. Org. Lett. 2017, 19, 5284–5287. [Google Scholar] [CrossRef]
- Yang, J.; Wang, Y.; Wu, S.; Chen, F.-X. The highly efficient 1,4-addition of TMSCN to aromatic enones catalyzed by CsF with water as the additive. Synlett 2009, 3365–3367. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. A 2008, A64. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Cryst. C 2015, C71. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| KCN a | Temperature (°C)/Time (h) | Methanol/Water/AcOH b | Yield of 2aa, % c | |
|---|---|---|---|---|
| 1 | 130 | 65/1 | 2000/130/150 | 64 |
| 2 | 260 | 65/0.5 | 2000/260/300 | 65 |
| 3 | 130 | 65/0.5 | 1000/130/150 | 95(87) d |
| 4 | 130 | 20/12 | 1000/130/150 | 58 |
| 5 | 130 | 65/0.5 | 1000/0/150 | 79 |
| 6 | 130 | 65/0.5 | 1000/130/195 e | 38 |
| 7 | 130 | 65/0.5 | 1000/130/92 f | 49 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aksenov, N.A.; Aksenov, A.V.; Kurenkov, I.A.; Kirillov, N.K.; Aksenov, D.A.; Arutiunov, N.A.; Aksenova, D.S.; Rubin, M. One-Pot Synthesis of (E)-2-(3-Oxoindolin-2-ylidene)-2-arylacetonitriles. Molecules 2022, 27, 2808. https://doi.org/10.3390/molecules27092808
Aksenov NA, Aksenov AV, Kurenkov IA, Kirillov NK, Aksenov DA, Arutiunov NA, Aksenova DS, Rubin M. One-Pot Synthesis of (E)-2-(3-Oxoindolin-2-ylidene)-2-arylacetonitriles. Molecules. 2022; 27(9):2808. https://doi.org/10.3390/molecules27092808
Chicago/Turabian StyleAksenov, Nicolai A., Alexander V. Aksenov, Igor A. Kurenkov, Nikita K. Kirillov, Dmitrii A. Aksenov, Nikolai A. Arutiunov, Daria S. Aksenova, and Michael Rubin. 2022. "One-Pot Synthesis of (E)-2-(3-Oxoindolin-2-ylidene)-2-arylacetonitriles" Molecules 27, no. 9: 2808. https://doi.org/10.3390/molecules27092808
APA StyleAksenov, N. A., Aksenov, A. V., Kurenkov, I. A., Kirillov, N. K., Aksenov, D. A., Arutiunov, N. A., Aksenova, D. S., & Rubin, M. (2022). One-Pot Synthesis of (E)-2-(3-Oxoindolin-2-ylidene)-2-arylacetonitriles. Molecules, 27(9), 2808. https://doi.org/10.3390/molecules27092808