
Dioscin-Mediated Autophagy Alleviates MPP+-Induced Neuronal Degeneration: An In Vitro Parkinson’s Disease Model

 ,
,  ,
, 
 ,
, 
Abstract
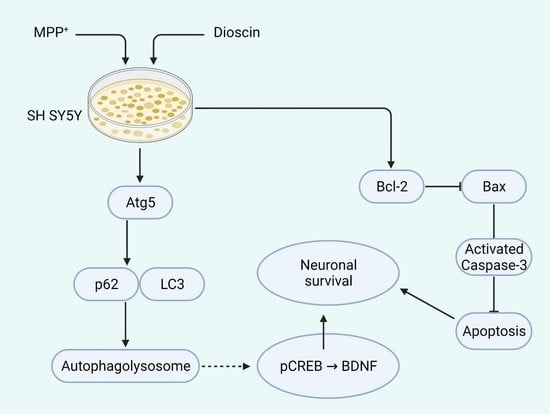
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Dioscin Protects against Neurotoxicity
2.2. Dioscin Dose-Dependently Downregulates Apoptotic Markers
2.3. Dioscin Dose-Dependently Increases TH Cells and Neurotrophic Factors
2.4. Dioscin Dose-Dependently Rescues Autophagic Function Impaired by MPP+
2.5. Dioscin Dose-Dependently Upregulates Autophagosome Formation
3. Discussion
4. Conclusions
5. Methods and Materials
5.1. Chemicals
5.2. Cell Culture and Treatment
5.3. Cell Viability Assay
5.4. Western Blot Analysis
5.5. Immunofluorescence
5.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
References
- Haque, M.E.; Akther, M.; Jakaria, M.; Kim, I.S.; Azam, S.; Choi, D.K. Targeting the Microglial NLRP3 Inflammasome and Its Role in Parkinson’s Disease. Mov. Disord. 2020, 35, 20–33. [Google Scholar] [CrossRef]
- Liu, J.; Wang, X.; Lu, Y.; Duan, C.; Gao, G.; Lu, L.; Yang, H. Pink1 interacts with α-synuclein and abrogates α-synuclein-induced neurotoxicity by activating autophagy. Cell Death Dis. 2017, 8, e3056. [Google Scholar] [CrossRef] [Green Version]
- Franco-Iborra, S.; Vila, M.; Perier, C. The Parkinson Disease Mitochondrial Hypothesis: Where Are We at? Neuroscientist 2016, 22, 266–277. [Google Scholar] [CrossRef]
- Jakaria, M.; Park, S.-Y.; Haque, M.E.; Karthivashan, G.; Kim, I.-S.; Ganesan, P.; Choi, D.-K. Neurotoxic Agent-Induced Injury in Neurodegenerative Disease Model: Focus on Involvement of Glutamate Receptors. Front. Mol. Neurosci. 2018, 11, 307. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef] [Green Version]
- Dauer, W.; Przedborski, S. Parkinson’s Disease: Mechanisms and Models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, A.M. Autophagy: Many paths to the same end. Mol. Cell. Biochem. 2004, 263, 55–72. [Google Scholar] [CrossRef]
- Kobayashi, S. Choose Delicately and Reuse Adequately: The Newly Revealed Process of Autophagy. Biol. Pharm. Bull. 2015, 38, 1098–1103. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.S.; Lin, L.; Geyer, A.; Haspel, J.A.; An, C.H.; Cao, J.; Rosas, I.O.; Morse, D. Autophagy in Idiopathic Pulmonary Fibrosis. PLoS ONE 2012, 7, e41394. [Google Scholar] [CrossRef]
- Levine, B.; Kroemer, G. Autophagy in the Pathogenesis of Disease. Cell 2008, 132, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Friedman, L.G.; Lachenmayer, M.L.; Wang, J.; He, L.; Poulose, S.M.; Komatsu, M.; Holstein, G.R.; Yue, Z. Disrupted Autophagy Leads to Dopaminergic Axon and Dendrite Degeneration and Promotes Presynaptic Accumulation of -Synuclein and LRRK2 in the Brain. J. Neurosci. 2012, 32, 7585–7593. [Google Scholar] [CrossRef] [Green Version]
- Prommahom, A.; Dharmasaroja, P. Effects of eEF1A2 knockdown on autophagy in an MPP+-induced cellular model of Parkinson’s disease. Neurosci. Res. 2021, 164, 55–69. [Google Scholar] [CrossRef]
- Ramalingam, M.; Kim, S.-J. Insulin on activation of autophagy with integrins and syndecans against MPP + -induced α-synuclein neurotoxicity. Neurosci. Lett. 2016, 633, 94–100. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, J.; Weng, L.; Li, X.; Yu, L.; Xu, Y. Valproic acid protects against MPP+-mediated neurotoxicity in SH-SY5Y Cells through autophagy. Neurosci. Lett. 2017, 638, 60–68. [Google Scholar] [CrossRef]
- Garcia-Garcia, A.; Anandhan, A.; Burns, M.; Chen, H.; Zhou, Y.; Franco, R. Impairment of Atg5-Dependent Autophagic Flux Promotes Paraquat- and MPP+-Induced Apoptosis But Not Rotenone or 6-Hydroxydopamine Toxicity. Toxicol. Sci. 2013, 136, 166–182. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, M.-J.; Tsai, T.-L.; Hsieh, Y.-S.; Wang, C.-J.; Chiou, H.-L. Dioscin-induced autophagy mitigates cell apoptosis through modulation of PI3K/Akt and ERK and JNK signaling pathways in human lung cancer cell lines. Arch. Toxicol. 2013, 87, 1927–1937. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Han, K.; Wang, C.; Sun, C.; Jia, N. Dioscin Protects against Aβ1–42 Oligomers-Induced Neurotoxicity via the Function of SIRT3 and Autophagy. Chem. Pharm. Bull. 2020, 68, 717–725. [Google Scholar] [CrossRef]
- Qi, Y.; Li, R.; Xu, L.; Yin, L.; Xu, Y.; Han, X.; Peng, J. Neuroprotective Effect of Dioscin on the Aging Brain. Molecules 2019, 24, 1247. [Google Scholar] [CrossRef] [Green Version]
- Du, S.; Li, C.; Lu, Y.; Lei, X.; Zhang, Y.; Li, S.; Liu, F.; Chen, Y.; Weng, D.; Chen, J. Dioscin Alleviates Crystalline Silica-Induced Pulmonary Inflammation and Fibrosis through Promoting Alveolar Macrophage Autophagy. Theranostics 2019, 9, 1878–1892. [Google Scholar] [CrossRef]
- Zhu, S.; Tang, S.; Su, F. Dioscin inhibits ischemic stroke-induced inflammation through inhibition of the TLR4/MyD88/NF-κB signaling pathway in a rat model. Mol. Med. Rep. 2019, 17, 660–666. [Google Scholar] [CrossRef]
- Yang, R.; Chen, W.; Lu, Y.; Li, Y.; Du, H.; Gao, S.; Dong, X.; Yuan, H. Dioscin relieves endotoxemia induced acute neuro-inflammation and protect neurogenesis via improving 5-HT metabolism. Sci. Rep. 2017, 7, 40035. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Du, Q.; Wang, F.; Liu, Z.; Li, B.; Wang, A.; Wang, Y. Microarray analysis of gene expression on herbal glycoside recipes improving deficient ability of spatial learning memory in ischemic mice. J. Neurochem. 2004, 88, 1406–1415. [Google Scholar] [CrossRef]
- Hu, Z.; Chen, B.; Zhang, J.-P.; Ma, Y.-Y. Up-regulation of autophagy-related gene 5 (ATG5) protects dopaminergic neurons in a zebrafish model of Parkinson’s disease. J. Biol. Chem. 2017, 292, 18062–18074. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Zhang, J.; Zhang, Q. Expression pattern and functions of autophagy-related geneatg5in zebrafish organogenesis. Autophagy 2011, 7, 1514–1527. [Google Scholar] [CrossRef] [Green Version]
- Khwanraj, K.; Phruksaniyom, C.; Madlah, S.; Dharmasaroja, P. Differential Expression of Tyrosine Hydroxylase Protein and Apoptosis-Related Genes in Differentiated and Undifferentiated SH-SY5Y Neuroblastoma Cells Treated with MPP+. Neurol. Res. Int. 2015, 2015, 734703. [Google Scholar] [CrossRef] [Green Version]
- Fan, F.; Li, S.; Wen, Z.; Ye, Q.; Chen, X.; Ye, Q. Regulation of PGC-1α mediated by acetylation and phosphorylation in MPP+ induced cell model of Parkinson’s disease. Aging 2020, 12, 9461–9474. [Google Scholar] [CrossRef]
- Kalivendi, S.V.; Cunningham, S.; Kotamraju, S.; Joseph, J.; Hillard, C.J.; Kalyanaraman, B. α-Synuclein Up-regulation and Aggregation during MPP+-induced Apoptosis in Neuroblastoma Cells. J. Biol. Chem. 2004, 279, 15240–15247. [Google Scholar] [CrossRef] [Green Version]
- Abd-Elrahman, K.S.; Ferguson, S.S.G. Modulation of mTOR and CREB pathways following mGluR5 blockade contribute to improved Huntington’s pathology in zQ175 mice. Mol. Brain 2019, 12, 35. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.; Zhou, X.-J.; Zhang, H. Exploring the Role of Autophagy-Related Gene 5 (ATG5) Yields Important Insights Into Autophagy in Autoimmune/Autoinflammatory Diseases. Front. Immunol. 2018, 9, 2334. [Google Scholar] [CrossRef]
- Lei, Z.; Cao, G.; Wei, G. A30P mutant α-synuclein impairs autophagic flux by inactivating JNK signaling to enhance ZKSCAN3 activity in midbrain dopaminergic neurons. Cell Death Dis. 2019, 10, 133. [Google Scholar] [CrossRef] [Green Version]
- Singer, T.P.; Ramsay, R. Mechanism of the neurotoxicity of MPTP. FEBS Lett. 1990, 274, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Eberhardt, O.; Schulz, J.B. Apoptotic mechanisms and antiapoptotic therapy in the MPTP model of Parkinson’s disease. Toxicol. Lett. 2003, 139, 135–151. [Google Scholar] [CrossRef]
- Lim, J.; Kim, H.-W.; Youdim, M.B.; Rhyu, I.J.; Choe, K.-M.; Oh, Y.J. Binding preference of p62 towards LC3-ll during dopaminergic neurotoxin-induced impairment of autophagic flux. Autophagy 2011, 7, 51–60. [Google Scholar] [CrossRef] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to Interpret LC3 Immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- Turmel, H.; Hartmann, A.; Parain, K.; Douhou, A.; Srinivasan, A.; Agid, Y.; Hirsch, E.C. Caspase-3 activation in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated mice. Mov. Disord. 2001, 16, 185–189. [Google Scholar] [CrossRef]
- Gómez, C.; Reiriz, J.; Piqué, M.; Gil, J.; Ferrer, I.; Ambrosio, S. Low concentrations of 1-methyl-4-phenylpyridinium ion induce caspase-mediated apoptosis in human SH-SY5Y neuroblastoma cells. J. Neurosci. Res. 2001, 63, 421–428. [Google Scholar] [CrossRef]
- Friedlander, R.M. Apoptosis and Caspases in Neurodegenerative Diseases. N. Engl. J. Med. 2003, 348, 1365–1375. [Google Scholar] [CrossRef]
- Carloni, S.; Girelli, S.; Scopa, C.; Buonocore, G.; Longini, M.; Balduini, W. Activation of autophagy and Akt/CREB signaling play an equivalent role in the neuroprotective effect of rapamycin in neonatal hypoxia-ischemia. Autophagy 2010, 6, 366–377. [Google Scholar] [CrossRef] [Green Version]
- Gandolfi, D.; Cerri, S.; Mapelli, J.; Polimeni, M.; Tritto, S.; Fuzzati-Armentero, M.-T.; Bigiani, A.; Blandini, F.; Mapelli, L.; D’Angelo, E. Activation of the CREB/c-Fos Pathway during Long-Term Synaptic Plasticity in the Cerebellum Granular Layer. Front. Cell. Neurosci. 2017, 11. [Google Scholar] [CrossRef] [Green Version]
- Saura, C.A.; Cardinaux, J.-R. Emerging Roles of CREB-Regulated Transcription Coactivators in Brain Physiology and Pathology. Trends Neurosci. 2017, 40, 720–733. [Google Scholar] [CrossRef] [Green Version]
- Tao, X.; Finkbeiner, S.; Arnold, D.B.; Shaywitz, A.J.; Greenberg, M.E. Ca2+ Influx Regulates BDNF Transcription by a CREB Family Transcription Factor-Dependent Mechanism. Neuron 1998, 20, 709–726. [Google Scholar] [CrossRef] [Green Version]
- Dong, M.; Wu, Y.; Fan, Y.; Xu, M.; Zhang, J. c-fos modulates brain-derived neurotrophic factor mRNA expression in mouse hippocampal CA3 and dentate gyrus neurons. Neurosci. Lett. 2006, 400, 177–180. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, D.; McQuade, J.S.; Behbehani, M.M.; Tsien, J.Z.; Xu, M. c-fos regulates neuronal excitability and survival. Nat. Genet. 2002, 30, 416–420. [Google Scholar] [CrossRef]
- Lipsky, R.H.; Marini, A.M. Brain-Derived Neurotrophic Factor in Neuronal Survival and Behavior-Related Plasticity. Ann. N. Y. Acad. Sci. 2007, 1122, 130–143. [Google Scholar] [CrossRef]
- Selvaraj, S.; Sun, Y.; Watt, J.A.; Wang, S.; Lei, S.; Birnbaumer, L.; Singh, B.B. Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J. Clin. Investig. 2012, 122, 1354–1367. [Google Scholar] [CrossRef] [Green Version]
- Nam, J.H.; Leem, E.; Jeon, M.-T.; Jeong, K.H.; Park, J.-W.; Jung, U.J.; Kholodilov, N.; Burke, R.E.; Jin, B.K.; Kim, S.R. Induction of GDNF and BDNF by hRheb(S16H) Transduction of SNpc Neurons: Neuroprotective Mechanisms of hRheb(S16H) in a Model of Parkinson’s Disease. Mol. Neurobiol. 2014, 51, 487–499. [Google Scholar] [CrossRef]
- Haque, M.E.; Akther, M.; Azam, S.; Choi, D.-K.; Kim, I.-S. GPR4 Knockout Improves the Neurotoxin-Induced, Caspase-Dependent Mitochondrial Apoptosis of the Dopaminergic Neuronal Cell. Int. J. Mol. Sci. 2020, 21, 7517. [Google Scholar] [CrossRef]
- Jakaria, M.; Azam, S.; Cho, D.-Y.; Haque, M.E.; Kim, I.-S.; Choi, D.-K. The Methanol Extract of Allium cepa L. Protects Inflammatory Markers in LPS-Induced BV-2 Microglial Cells and Upregulates the Antiapoptotic Gene and Antioxidant Enzymes in N27-A Cells. Antioxidants 2019, 8, 348. [Google Scholar] [CrossRef] [Green Version]
- Runwal, G.; Stamatakou, E.; Siddiqi, F.H.; Puri, C.; Zhu, Y.; Rubinsztein, D.C. LC3-positive structures are prominent in autophagy-deficient cells. Sci. Rep. 2019, 9, 10147. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azam, S.; Haque, M.E.; Cho, D.-Y.; Kim, J.-S.; Jakaria, M.; Kim, I.-S.; Choi, D.-K. Dioscin-Mediated Autophagy Alleviates MPP+-Induced Neuronal Degeneration: An In Vitro Parkinson’s Disease Model. Molecules 2022, 27, 2827. https://doi.org/10.3390/molecules27092827
Azam S, Haque ME, Cho D-Y, Kim J-S, Jakaria M, Kim I-S, Choi D-K. Dioscin-Mediated Autophagy Alleviates MPP+-Induced Neuronal Degeneration: An In Vitro Parkinson’s Disease Model. Molecules. 2022; 27(9):2827. https://doi.org/10.3390/molecules27092827
Chicago/Turabian StyleAzam, Shofiul, Md. Ezazul Haque, Duk-Yeon Cho, Joon-Soo Kim, Md. Jakaria, In-Su Kim, and Dong-Kug Choi. 2022. "Dioscin-Mediated Autophagy Alleviates MPP+-Induced Neuronal Degeneration: An In Vitro Parkinson’s Disease Model" Molecules 27, no. 9: 2827. https://doi.org/10.3390/molecules27092827
APA StyleAzam, S., Haque, M. E., Cho, D.-Y., Kim, J.-S., Jakaria, M., Kim, I.-S., & Choi, D.-K. (2022). Dioscin-Mediated Autophagy Alleviates MPP+-Induced Neuronal Degeneration: An In Vitro Parkinson’s Disease Model. Molecules, 27(9), 2827. https://doi.org/10.3390/molecules27092827