


Asymmetric Organocatalysis: A Survival Guide to Medicinal Chemists †


{kind=link}
{kind=link}
{kind=link}
{kind=link}
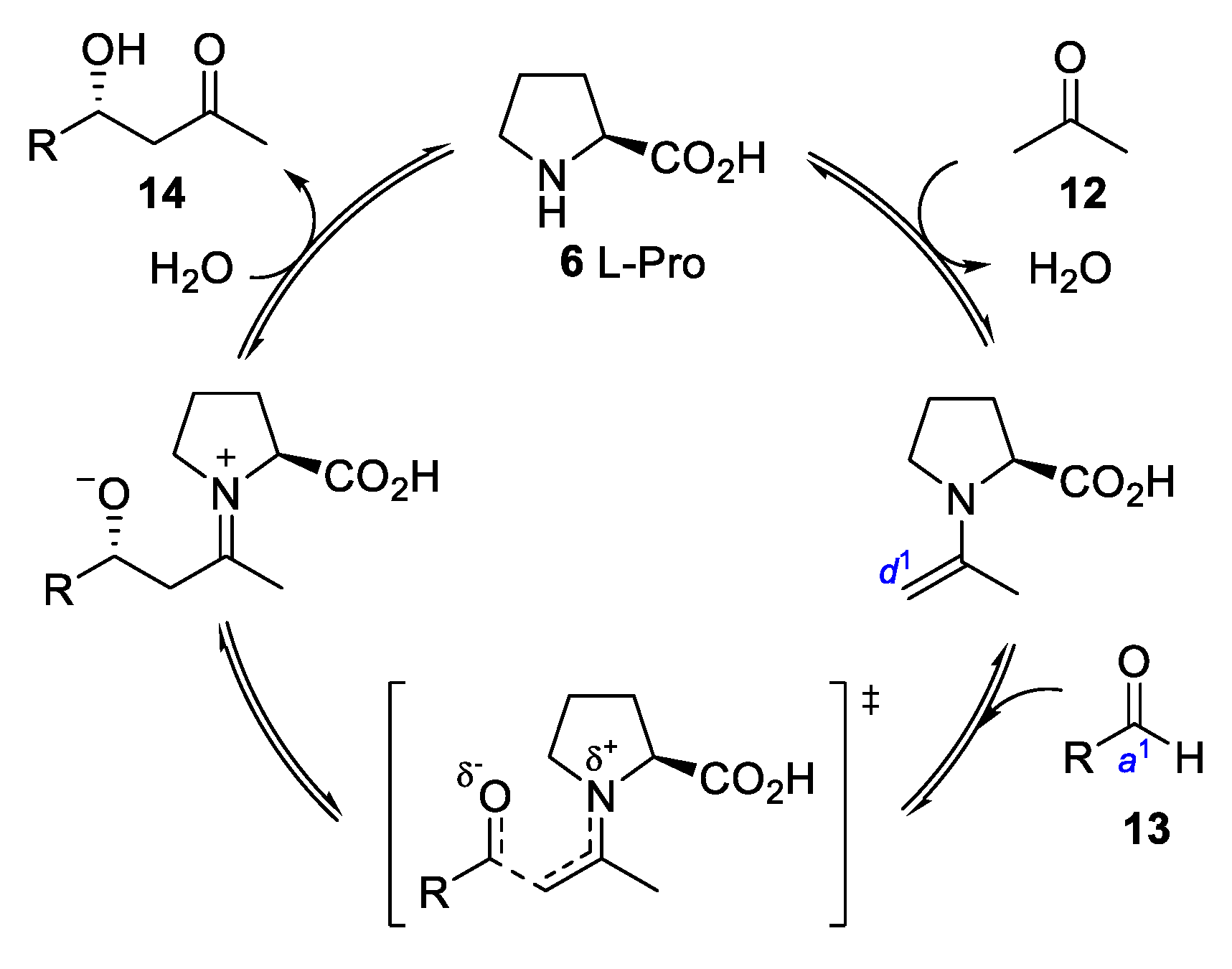{kind=link}
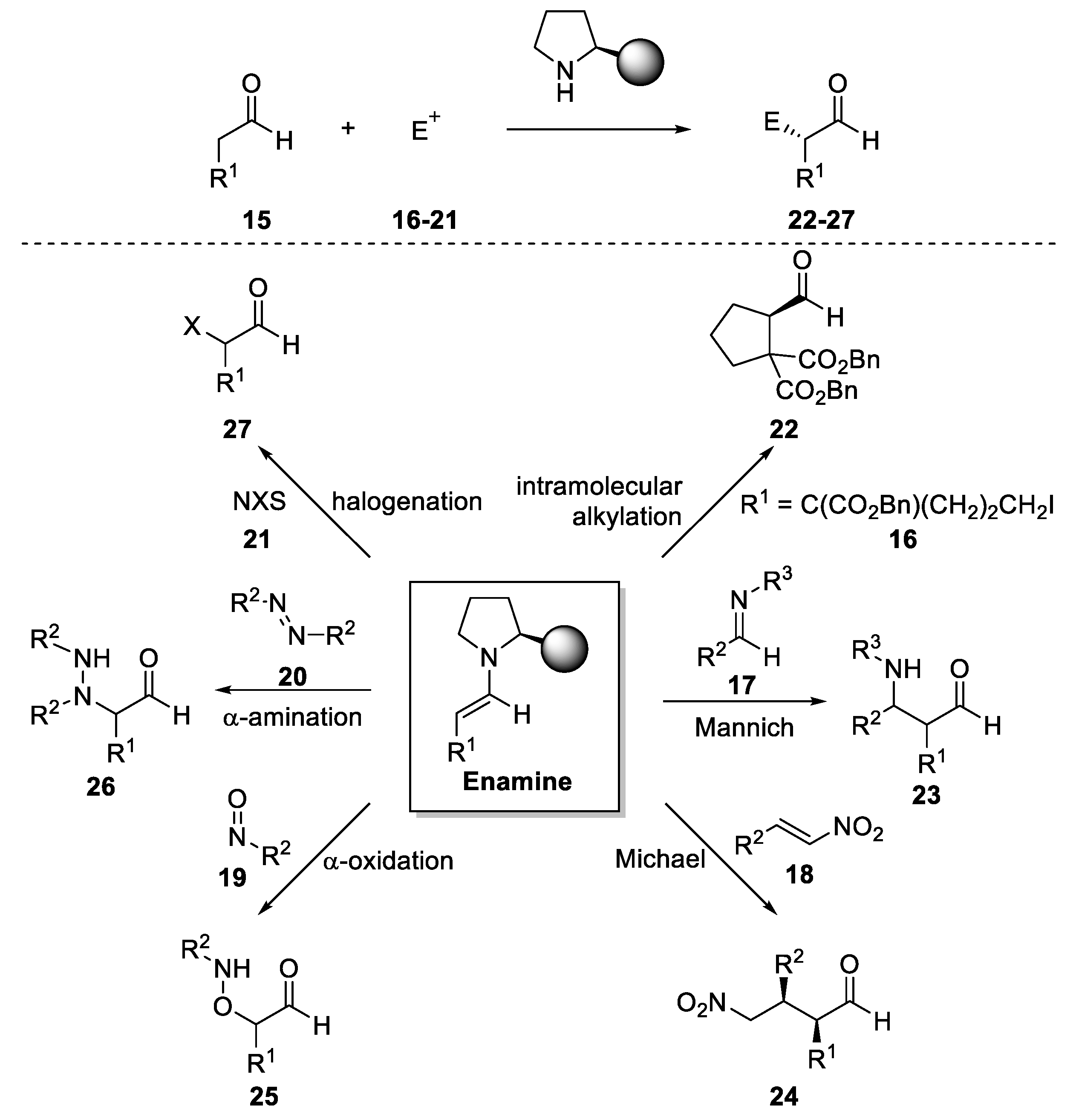{kind=link}
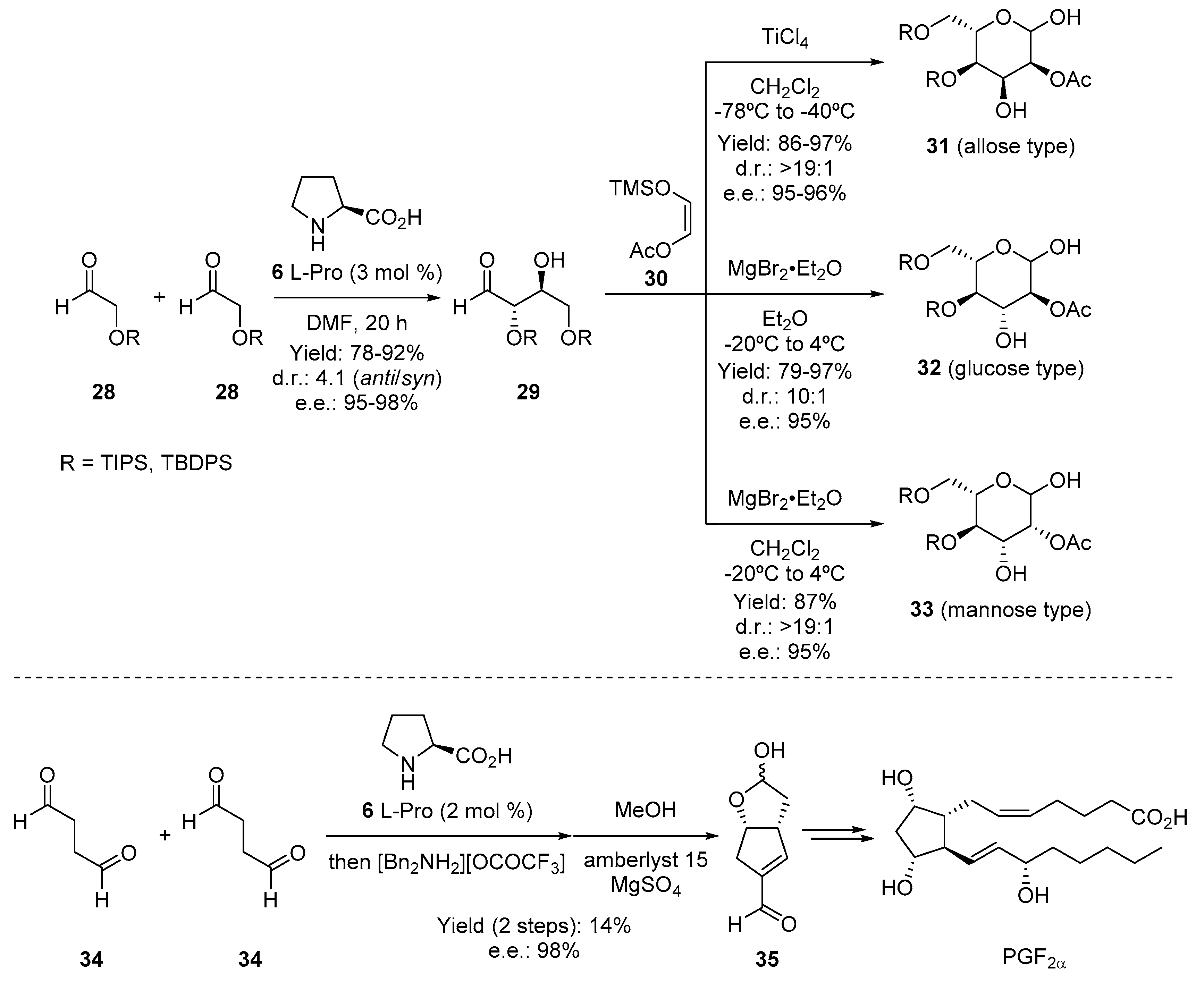{kind=link}
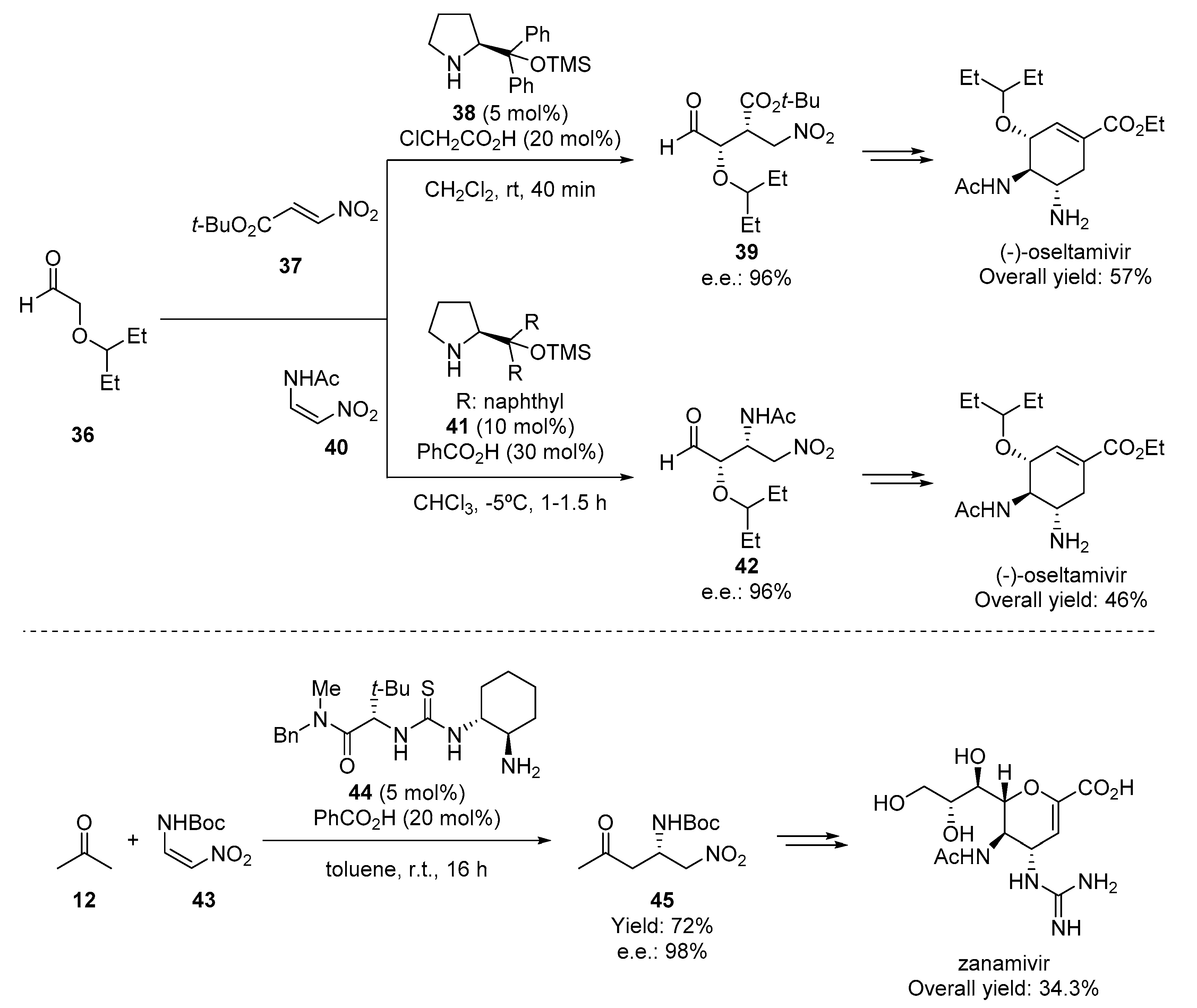{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Generic Activation Modes
3. Enamine Catalysis
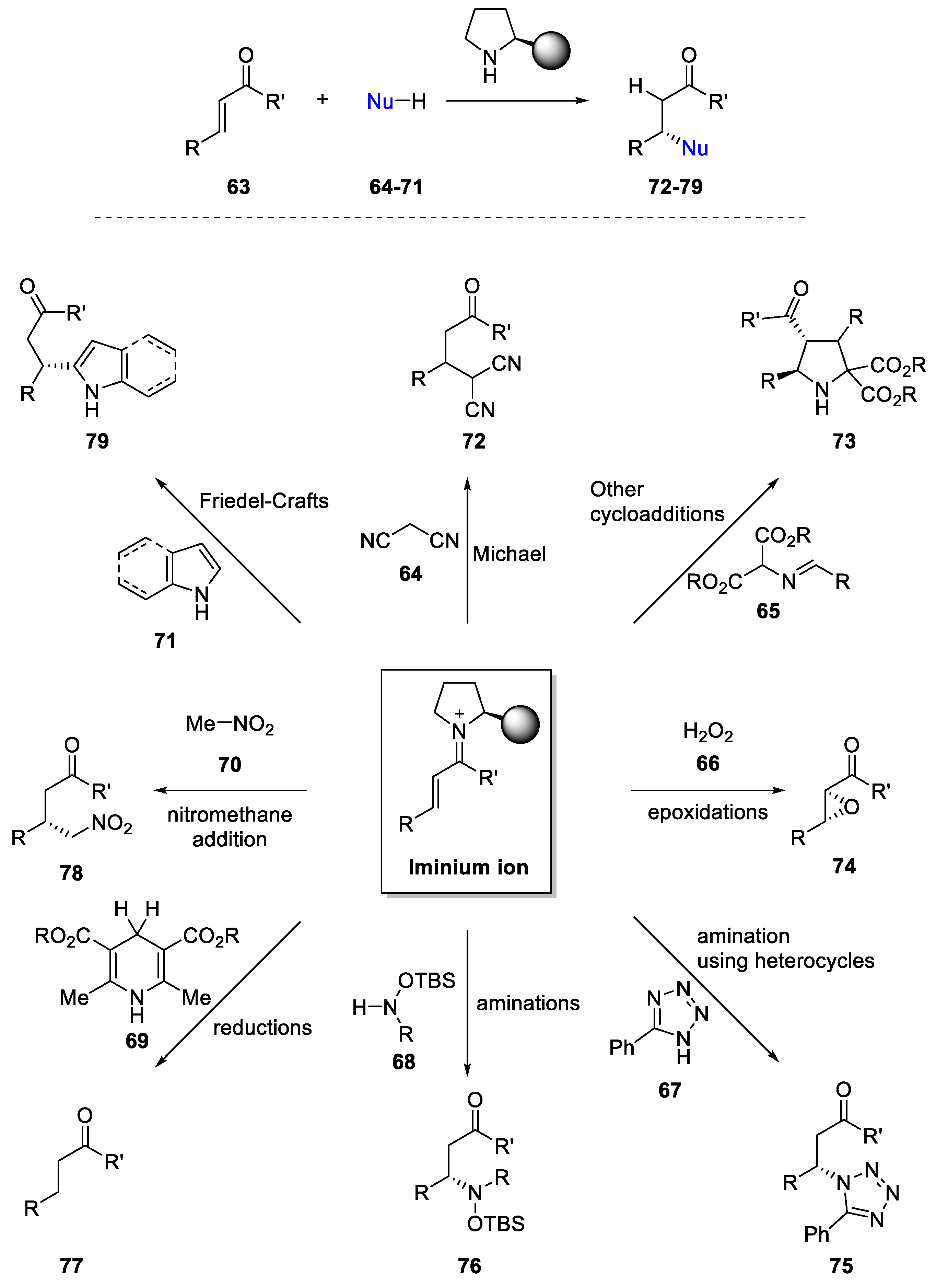
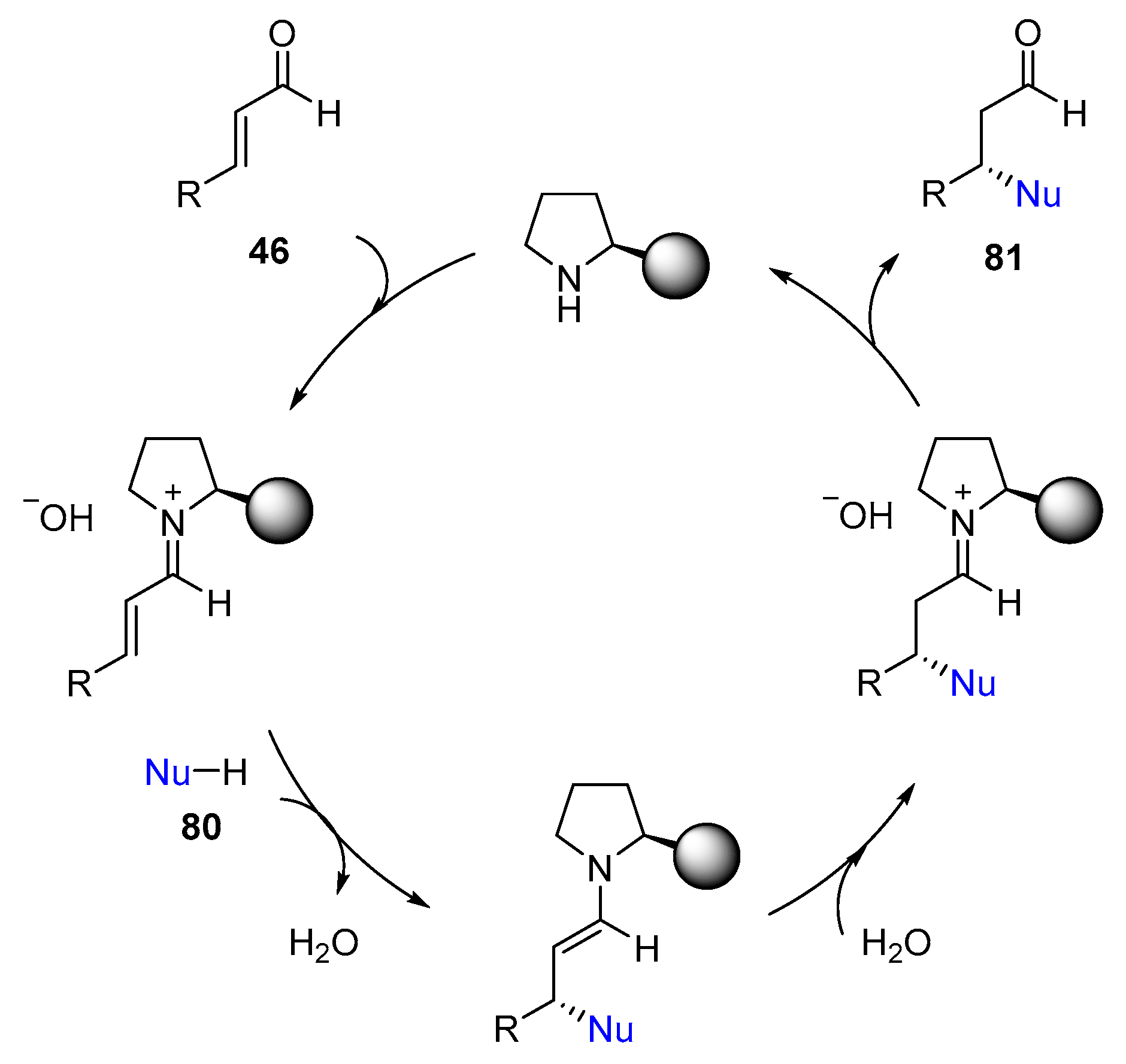
4. Iminium-Ion Catalysis
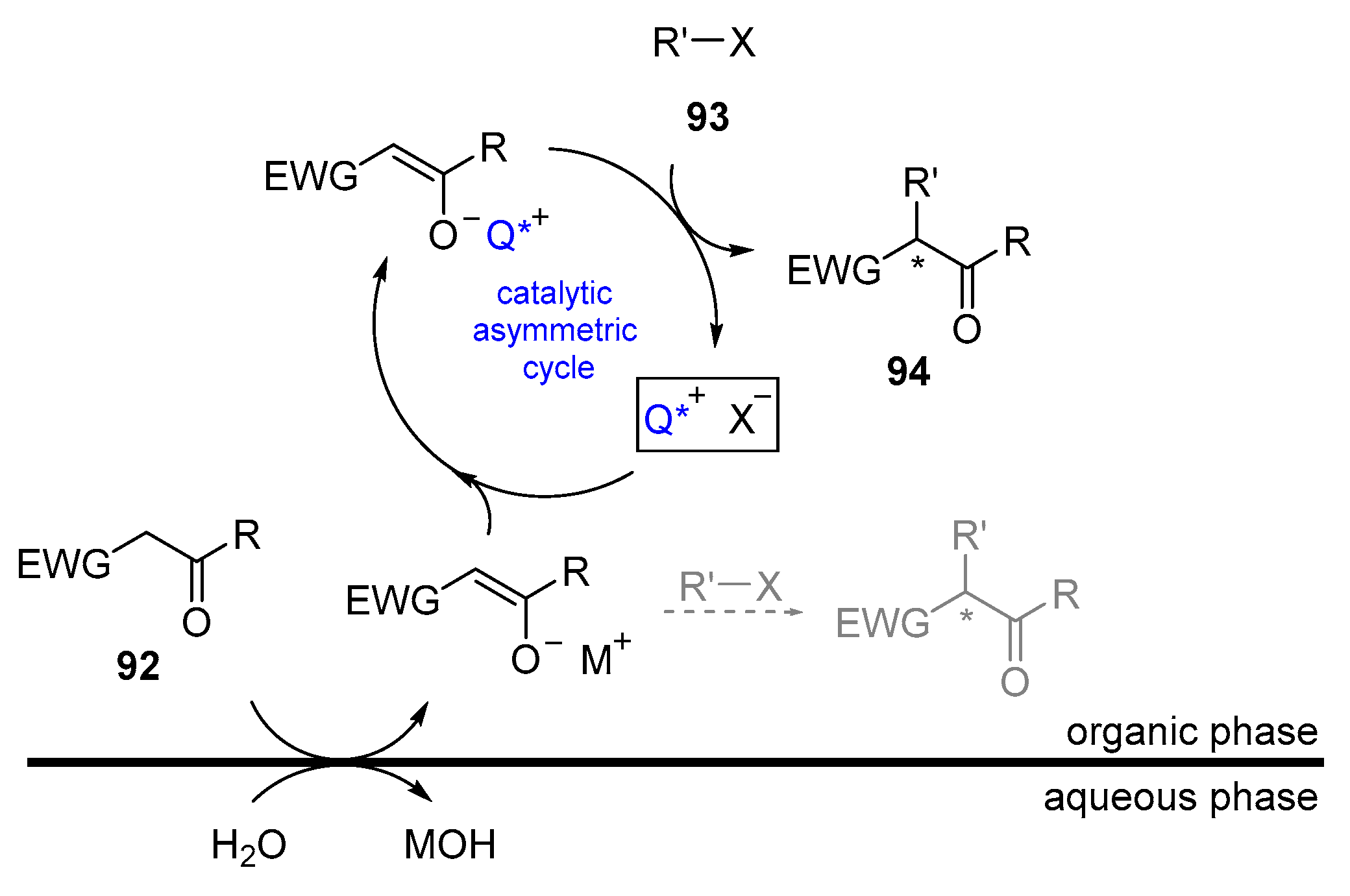
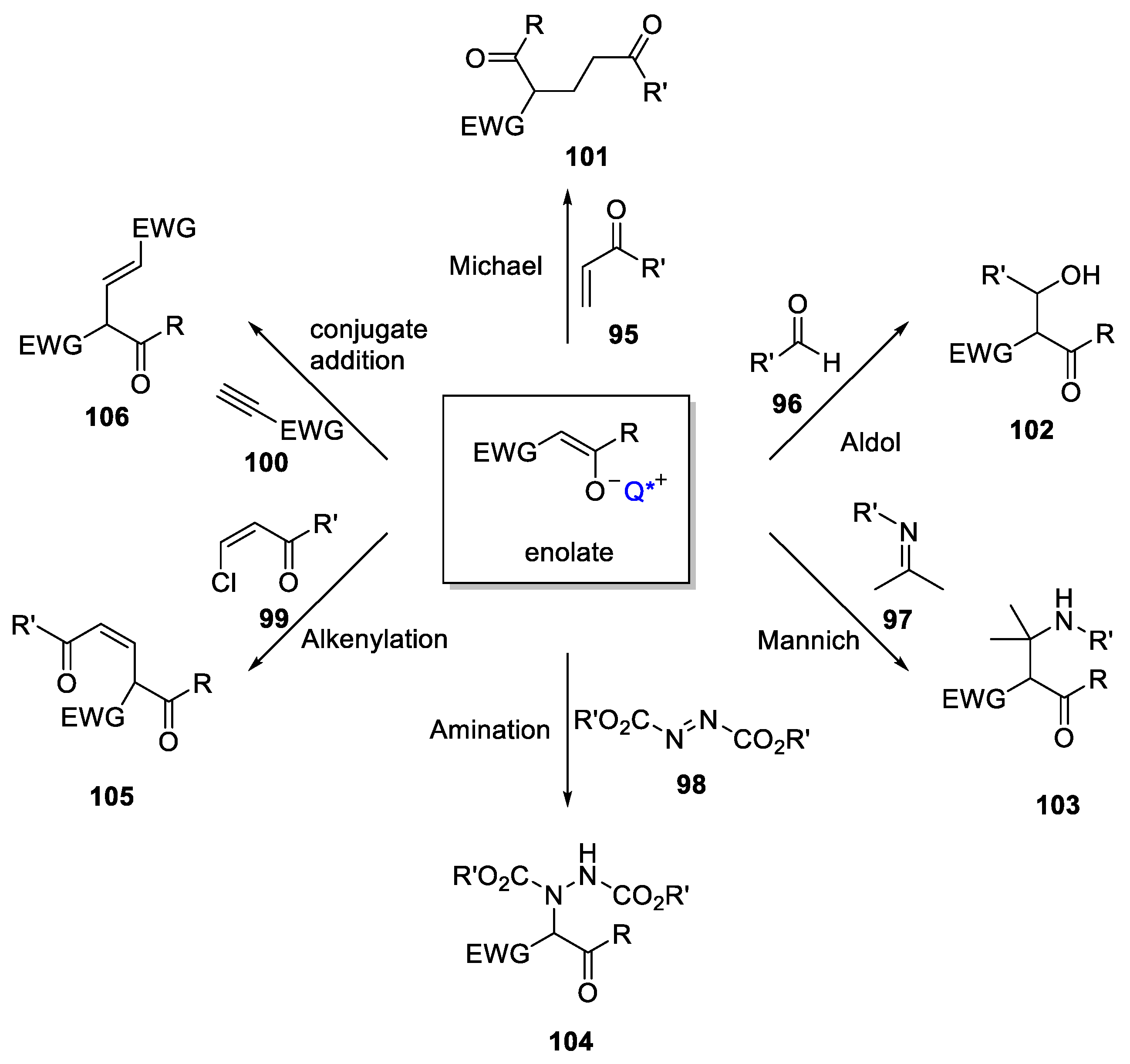
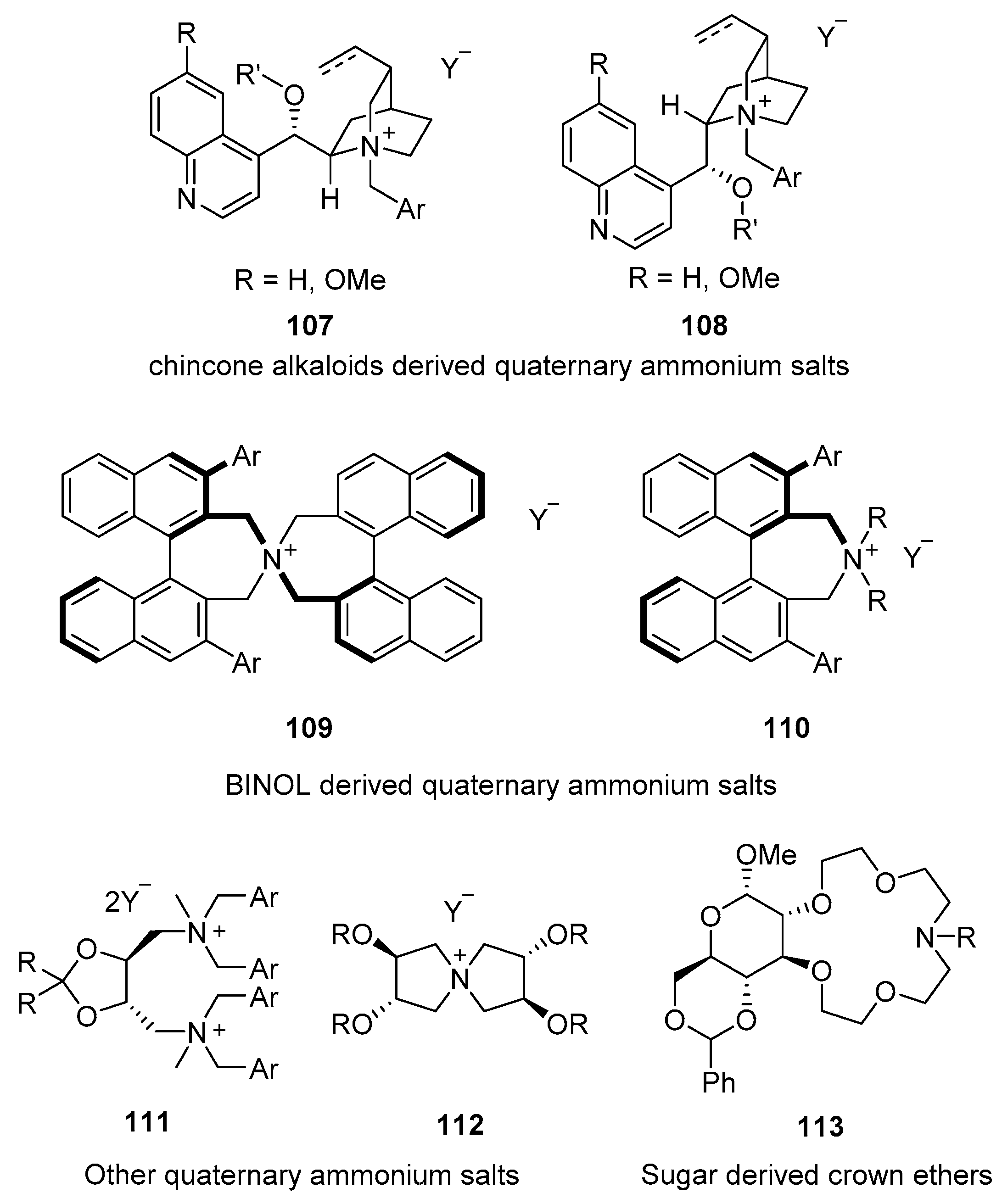
5. PTC Catalysis
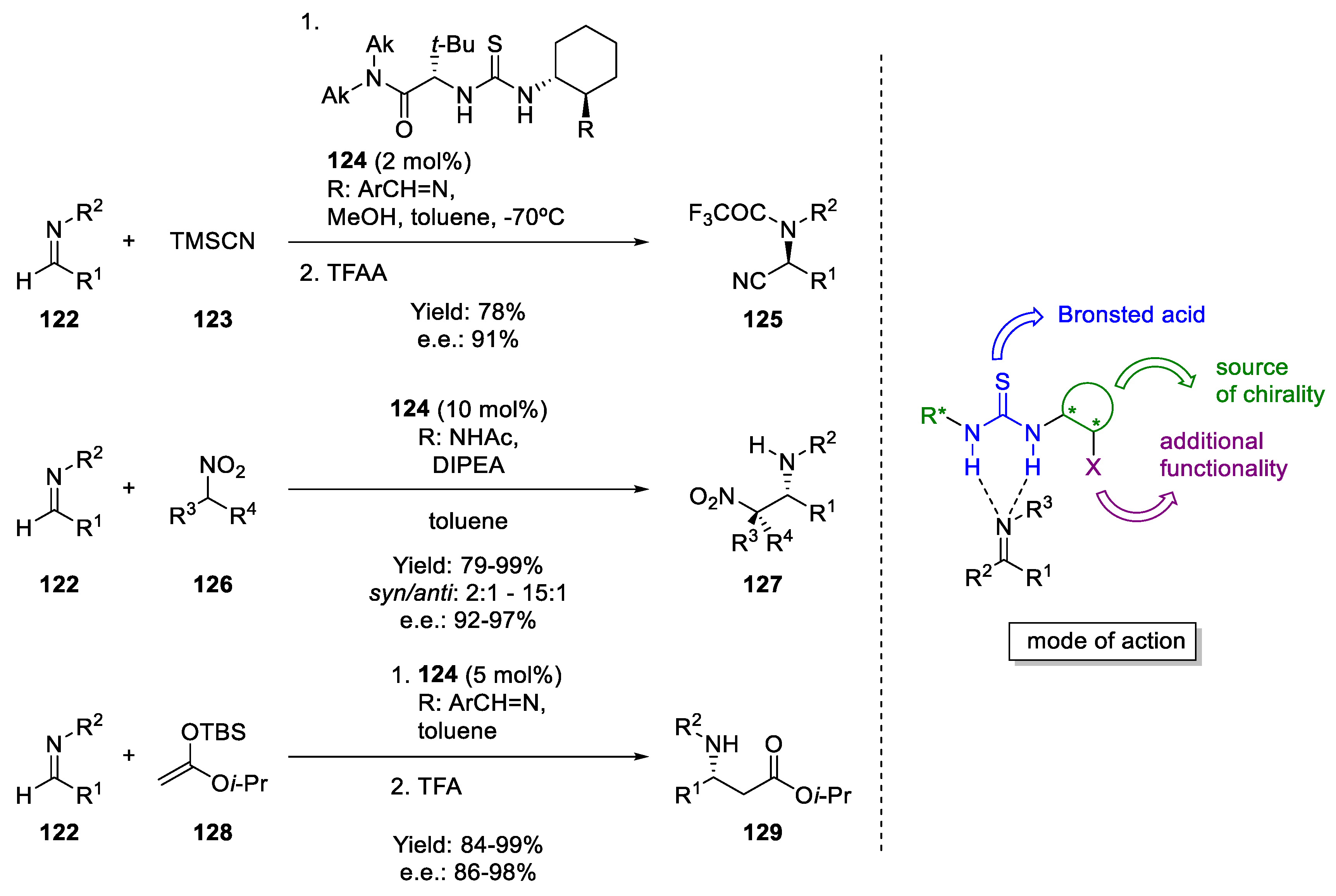
6. H-Bond Catalysis
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- ReferencesAgranat, I.; Caner, H.; Caldwell, J. Putting chirality to work: The strategy of chiral switches. Nat. Rev. Drug Discov. 2002, 1, 753–768. [Google Scholar]
- Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-new-stereoisomeric-drugs (accessed on 15 December 2022).
- Agranat, I.; Wainschtein, S.R.; Zusman, E.Z. The predicated demise of racemic new molecular entities is an exaggeration. Nat. Rev. Drug Discov. 2012, 11, 972–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seebach, D.; Hungerbühler, E. Synthesis of Enantiomerically Pure Compounds (EPC-Synthesis). In Modern Synthetic Methods; Scheffold, R., Ed.; Salle + Sauerländer: Frankfurt, Germany, 1980. [Google Scholar]
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Ahrendt, K.A.; Borths, C.J.; MacMillan, D.W.C. New strategies for organic catalysis: The first highly enantioselective organocatalytic Diels—Alder reaction. J. Am. Chem. Soc. 2000, 122, 4243–4244. [Google Scholar] [CrossRef]
- List, B.; Lerner, R.A.; Barbas, C.F. Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Bredig, G.; Fiske, P.S. Durch Katalysatoren bewirkte Asymmetrische Synthese. Biochem. Z. 1912, 687, 7–23. [Google Scholar]
- Hajos, Z.G.; Parrish, D.R. Asymmetric synthesis of bicyclic intermediates of natural product chemistry. J. Org. Chem. 1974, 39, 1615–1621. [Google Scholar] [CrossRef]
- Eder, U.; Sauer, G.; Wiecert, R. New Type of Asymmetric Cyclization to Optically Active Steroid CD Partial Structures. Angew. Chem. Int. Ed. 1971, 10, 496–497. [Google Scholar] [CrossRef]
- Sigman, M.S.; Jacobsen, E.N. Schiff Base Catalysts for the Asymmetric Strecker Reaction Identified and Optimized from Parallel Synthetic Libraries. J. Am. Chem. Soc. 1998, 120, 4901–4902. [Google Scholar] [CrossRef]
- Zhong, G.; Lerner, R.A.; Barbas, C.F. Broadening the Aldolase Catalytic Antibody Repertoire by Combining Reactive Immunization and Transition State Theory: New Enantio- and Diastereoselectivities. Angew. Chem. Int. Ed. 1999, 38, 3738–3741. [Google Scholar] [CrossRef]
- List, B.; Lerner, R.A.; Barbas, C.F., III. Enantioselective Aldol Cyclodehydrations Catalyzed by Antibody 38C2. Org. Lett. 1999, 1, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Zhong, G.; Hoffmann, T.; Lerner, R.A.; Danishefsky, S.; Barbas, C.F., III. Antibody-Catalyzed Enantioselective Robinson Annulation. J. Am. Chem. Soc. 1997, 119, 8131–8132. [Google Scholar] [CrossRef]
- Barbas, C.F. Organocatalysis Lost: Modern Chemistry, Ancient Chemistry, and an Unseen Biosynthetic Apparatus. Angew. Chem. Int. Ed. 2007, 47, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; He, X.-H.; Liu, Y.-Q.; He, G.; Peng, C.; Li, J.-L. Asymmetric organocatalysis: An enabling technology for medicinal chemistry. Chem. Soc. Rev. 2021, 50, 1522–1586. [Google Scholar] [CrossRef] [PubMed]
- Alemán, J.; Cabrera, S. Applications of Asymmetric Organocatalysis in Medicinal Chemistry. Chem. Soc. Rev. 2013, 42, 774–793. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, L.; Albrecht, A.; Dell’Amico, L. (Eds.) Asymmetric Organocatalysis: New Strategies, Catalysts, and Opportunities; Wiley-VCH: Weinheim, Germany, 2022. [Google Scholar]
- Reyes-Rodriguez, G.J.; Rezayee, N.M.; Vidal-Albalat, A.; Jørgensen, K.A. Prevalence of Diarylprolinol Silyl Ethers as Catalysts in Total Synthesis and Patents. Chem. Rev. 2019, 119, 4221–4260. [Google Scholar] [CrossRef]
- Helm, M.P.v.d.; Klemm, B.; Eelkema, R. Organocatalysis in aqueous media. Nat. Rev. Chem. 2019, 3, 491–508. [Google Scholar] [CrossRef] [Green Version]
- Sinibaldi, A.; Nori, V.; Baschieri, A.; Fini, F.; Arcadi, A.; Carlone, A. Organoca-talysis and Beyond: Activating Reactions with Two Catalytic Species. Catalysts 2019, 9, 928. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, V.d.G.; Cardoso, M.F.d.C.; Forezi, L.d.S.M. Organocatalysis: A Brief Overview on Its Evolution and Applications. Catalysts 2018, 8, 605. [Google Scholar] [CrossRef] [Green Version]
- Dalko, P.I. (Ed.) Comprehensive Enantioselective Organocatalysis: Catalysts, Re-Actions, and Applications; Wiley-VCH: Weinheim, Germany, 2013. [Google Scholar]
- Jacobsen, E.N.; MacMillan, D.W.C. Organocatalysis. Proc. Natl. Acad. Sci. USA 2010, 107, 20618–20619. [Google Scholar] [CrossRef] [Green Version]
- Bertelsen, S.; Jørgensen, K.A. Organocatalysis—after the gold rush. Chem. Soc. Rev. 2009, 38, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- Dondoni, A.; Massi, A. Asymmetric Organocatalysis: From Infancy to Adolescence. Angew. Chem. Int. Ed. 2008, 47, 4638–4660. [Google Scholar] [CrossRef] [PubMed]
- List, B.; Yang, J.W. The Organic Approach to Asymmetric Catalysis. Science 2006, 313, 1584–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berkessel, A. (Ed.) Asymmetric Organocatalysis: From Biomi-Metic Concepts to Applications in Asymmetric Synthesis; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- List, B.; Houk, K.N. Aymmetric Organocatalysis. Acc. Chem. Res. 2004, 37, 487. [Google Scholar]
- Dalko, P.I.; Moisan, L. In the Golden Age of Organocatalysis. Angew. Chem. Int. Ed. 2004, 43, 5138–5175. [Google Scholar] [CrossRef]
- Dalko, P.I.; Moisan, L. Enantioselective Organocatalysis. Angew. Chem. Int. Ed. 2001, 40, 3726–3748. [Google Scholar] [CrossRef]
- Holland, M.C.; Gilmour, R. Deconstructing Covalent Organocatalysis. Angew. Chem. Int. Ed. 2015, 54, 3862–3871. [Google Scholar] [CrossRef]
- Belloti, P.; Koy, M.; Hopkinson, M.N.; Glorius, F. Recent advances in the chemistry and applications of N-heterocyclic carbenes. Nat. Rev. Chem. 2021, 5, 711–725. [Google Scholar] [CrossRef]
- Reyes, E.; Uria, U.; Carrillo, L.; Vicario, J.L. Enantioselective Cascade Reactions under N-Heterocyclic Carbene Catalysis. Synthesis 2017, 49, 451–471. [Google Scholar] [CrossRef]
- Flanigan, D.M.; Romano-Michailidis, F.; White, N.A.; Rovis, T. Organocatalytic Reactions Enabled by N-Heterocyclic Carbenes. Chem. Rev. 2015, 115, 9307–9387. [Google Scholar] [CrossRef] [Green Version]
- Menon, R.S.; Biju, A.T.; Nair, V. Recent advances in employing homoenolates generated by N-heterocyclic carbene (NHC) catalysis in carbon–carbon bond-forming reactions. Chem. Soc. Rev. 2015, 44, 5040–5052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, S.J.; Candish, L.; Lupton, D.W. Acyl Anion free N-Heterocyclic Carbene Organocatalysis. Chem. Soc. Rev. 2013, 42, 4906. [Google Scholar] [CrossRef] [PubMed]
- Izquierdo, J.; Hutson, G.E.; Cohen, D.T.; Scheidt, K.A. A Continuum of Progress: Applications of N-Hetereocyclic Carbene Catalysis in Total Synthesis. Angew. Chem. Int. Ed. 2012, 51, 11686–11698. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Li, M.; Yuan, L.; Wang, J. Recent advances in heterocyclic carbene catalyzed achiral synthesis. Org. Bio. Chem. 2017, 15, 4731–4749. [Google Scholar] [CrossRef]
- Enders, D.; Niemeier, O.; Henseler, A. Organocatalysis by N-Heterocyclic Carbenes. Chem. Rev. 2007, 107, 5606. [Google Scholar] [CrossRef] [PubMed]
- Khong, S.; Venkatesh, T.; Kwon, O. Nucleophilic Phosphine Catalysis: The Untold Story. Asian J. Org. Chem. 2021, 10, 2699–2708. [Google Scholar] [CrossRef]
- Guo, H.; Fan, Y.C.; Sun, Z.; Wu, Y.; Kwon, O. Phosphine Organocatalysis. Chem. Rev. 2018, 118, 10049–10293. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Peng, B.; Ye, S.; Yin, Z.; Cao, J.; Xiao, X.; Zhao, B. Catalytic asymmetric α C(sp3)–H addition of benzylamines to aldehydes. Nat. Catal. 2022, 5, 1061–1068. [Google Scholar] [CrossRef]
- Chen, J.; Gong, X.; Li, J.; Li, Y.; Ma, J.; Hou, C.; Zhao, G.; Yuan, W.; Zhao, B. Carbonyl catalysis enables abiomimetic asymmetricMannich reaction. Chem. Rev. 2019, 119, 12033–12088. [Google Scholar]
- Parra, A. Chiral Hypervalent Iodines: Active Players in Asymmetric Synthesis. Chem. Rev. 2019, 119, 12033–12088. [Google Scholar] [CrossRef]
- Proctor, R.S.J.; Colgan, A.C.; Phipps, R.J. Exploiting attractive non-covalent interactions for the enantioselective catalysis of reactions involving radical intermediates. Nat. Chem. 2020, 12, 990–1004. [Google Scholar] [CrossRef] [PubMed]
- Žabka, M.; Šebesta, R. Experimental and Theoretical Studies in Hydrogen-Bonding Organocatalysis. Molecules 2015, 20, 15500–15524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, A.G.; Jacobsen, E.N. Small-Molecule H-Bond Donors in Asymmetric Catalysis. Chem. Rev. 2007, 107, 5713–5743. [Google Scholar] [CrossRef] [PubMed]
- Merad, J.; Lalli, C.; Bernadat, G.; Maury, J.; Masson, G. Enantioselective Brønsted Acid Catalysis as a Tool for the Synthesis of Natural Products and Pharmaceuticals. Chem. A. Eur. J. 2018, 24, 3925–3943. [Google Scholar] [CrossRef]
- Min, C.; Seidel, D. Asymmetric Brønsted acid catalysis with chiral carboxylic acids. Chem. Soc. Rev. 2017, 46, 5889–5902. [Google Scholar] [CrossRef]
- Cheon, C.H.; Yamamoto, H. Super Brønsted acid catalysis. Chem. Commun. 2011, 47, 3043–3056. [Google Scholar] [CrossRef]
- Kampen, D.; Reisinger, C.M.; List, B. Chiral Brønsted Acids for Asymmetric Organocatalysis. Top. Curr. Chem. 2010, 291, 395–456. [Google Scholar]
- Akiyama, T. Stronger Brønsted Acids. Chem. Rev. 2007, 107, 5744–5758. [Google Scholar] [CrossRef]
- Akiyama, T.; Itoh, J.; Fuchibe, K. Recent Progress in Chiral Brønsted Acid Catalysis. Adv. Synth. Catal. 2006, 348, 999–1010. [Google Scholar] [CrossRef]
- Shirakawa, S.; Maruoka, K. Recent Developments in Asymmetric Phase-Transfer Reactions. Angew. Chem. Int. Ed. 2013, 52, 4312–4348. [Google Scholar] [CrossRef]
- Maruoka, K. Practical Aspects of Recent Asymmetric Phase-Transfer Catalysis. Org. Process Res. Dev. 2008, 12, 679–697. [Google Scholar] [CrossRef]
- Ooi, T.; Maruoka, K. Recent Advances in Asymmetric Phase-Transfer Catalysis. Angew. Chem. Int. Ed. 2007, 46, 4222–4226. [Google Scholar] [CrossRef] [PubMed]
- Yoon, T.P.; Jacobsen, E.N. Privileged Chiral Catalysts. Science 2003, 299, 1691–1693. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, T.E.; König, B. Ion-Pairing Catalysis in Stereoselective, Light-Induced Transformations. J. Am. Chem. Soc. 2022, 144, 19207–19218. [Google Scholar] [CrossRef]
- Qian, D.; Sun, J. Recent Progress in Asymmetric Ion-Pairing Catalysis with Ammonium Salts. Chem. Eur. J. 2019, 15, 3740–3751. [Google Scholar] [CrossRef]
- Brak, K.; Jacobsen, E.N. Asymmetric Ion-Pairing Catalysis. Angew. Chem. Int. Ed. 2012, 52, 534–561. [Google Scholar] [CrossRef] [Green Version]
- Stork, G.; Terrell, R.; Szmuszkovicz, J. A New Synthesis of 2-Alkyl and 2-Acyl Ketones. J. Am. Chem. Soc. 1954, 76, 2029–2030. [Google Scholar] [CrossRef]
- Bahmanyar, S.; Houk, K.N. The Origin of Stereoselectivity in Proline-Catalyzed Intramolecular Aldol Reactions. J. Am. Chem. Soc. 2001, 123, 12911–12912. [Google Scholar] [CrossRef]
- Cohen, N. Asymmetric Induction in 19-Norsteroid Total Synthesis. Acc. Chem. Res. 1976, 9, 412–417. [Google Scholar] [CrossRef]
- Agami, C.; Levisalles, J.; Puchot, C. A New Diagnostic Tool for Elucidating the Mechanism of Enantioselective Reactions. Application to the Hajos-Parrish Reaction. J. Chem. Soc. Chem. Commun. 1985, 12, 2499. [Google Scholar] [CrossRef]
- Mukherjee, S.; Yang, J.W.; Hoffmann, S.; List, B. Asymmetric Enamine Catalysis. Chem. Rev. 2007, 107, 5471–5569. [Google Scholar] [CrossRef] [PubMed]
- Northrup, A.B.; MacMillan, D.W.C. Two-step synthesis of carbohydrates by selective aldol reactions. Science 2004, 305, 1752–1755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Northrup, A.B.; MacMillan, D.W.C. The First Direct and Enantioselective Cross-Aldol Reaction of Aldehydes. J. Am. Chem. Soc. 2002, 124, 6798–6799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coulthard, G.; Erb, W.; Aggarwal, V.K. Stereocontrolled organocatalytic synthesis of prostaglandin PGF2α in seven steps. Nature 2012, 489, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Yu, S.; Wang, Y.; Ma, D. Organocatalytic Michael Addition of Aldehydes to Protected 2-Amino-1-Nitroethenes: The Practical Syntheses of Oseltamivir (Tamiflu) and Substituted 3-Aminopyrrolidines. Angew. Chem. Int. Ed. 2010, 49, 4656–4660. [Google Scholar] [CrossRef]
- Ishikawa, H.; Suzuki, T.; Hayashi, Y. High-Yielding Synthesis of the Anti-Influenza Neuramidase Inhibitor (-)-Oseltamivir by Three “One-Pot” Operations. Angew. Chem. Int. Ed. 2009, 48, 1304–1307. [Google Scholar] [CrossRef]
- Tian, J.; Zhong, J.; Li, Y.; Ma, D. Organocatalytic and Scalable Synthesis of the Anti-Influenza Drugs Zanamivir, Laninamivir, and CS-8958. Angew. Chem. Int. Ed. 2014, 53, 13885–13888. [Google Scholar] [CrossRef]
- Donslund, B.S.; Johansen, T.K.; Poulsen, P.H.; Halskov, K.S.; Jørgensen, K.A. The Diarylprolinol Silyl Ethers: Ten Years After. Angew. Chem. Int. Ed. 2015, 54, 13860–13874. [Google Scholar] [CrossRef]
- Mukherjee, S.; Biswas, B. Organo-Cascade Catalysis: Application of Merged Iminium-Enamine Activation Technique and Related Cascade Reactivities. ChemistrySelect 2020, 5, 10704–10726. [Google Scholar] [CrossRef]
- Hanessian, S.; Chattopadhyay, K. Iminium Ion-Enamine Cascade Cyclizations: Facile Access to Structurally Diverse Azacyclic Compounds and Natural Products. Org. Lett. 2014, 16, 232–235. [Google Scholar] [CrossRef]
- Grondal, C.; Jeanty, M.; Enders, D. Organocatalytic cascade reactions as a new tool in total synthesis. Nat. Chem. 2010, 2, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Ozboya, K.E.; Rovis, T. Enamine/carbene cascade catalysis in the diastereo- and enantioselective synthesis of functionalized cyclopentanones. Chem. Sci. 2011, 2, 1835–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hepburn, H.B.; Dell’Amico, L.; Melchiorre, P. Enantioselective Vinylogous Organocascade Reactions. Chem. Rec. 2016, 16, 1787–1806. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Kumar, S.; Wang, H. Synergistic—Cooperative combination of enamine catalysis with transition metal catalysis. Chem. Commun. 2014, 50, 4272–4284. [Google Scholar] [CrossRef] [PubMed]
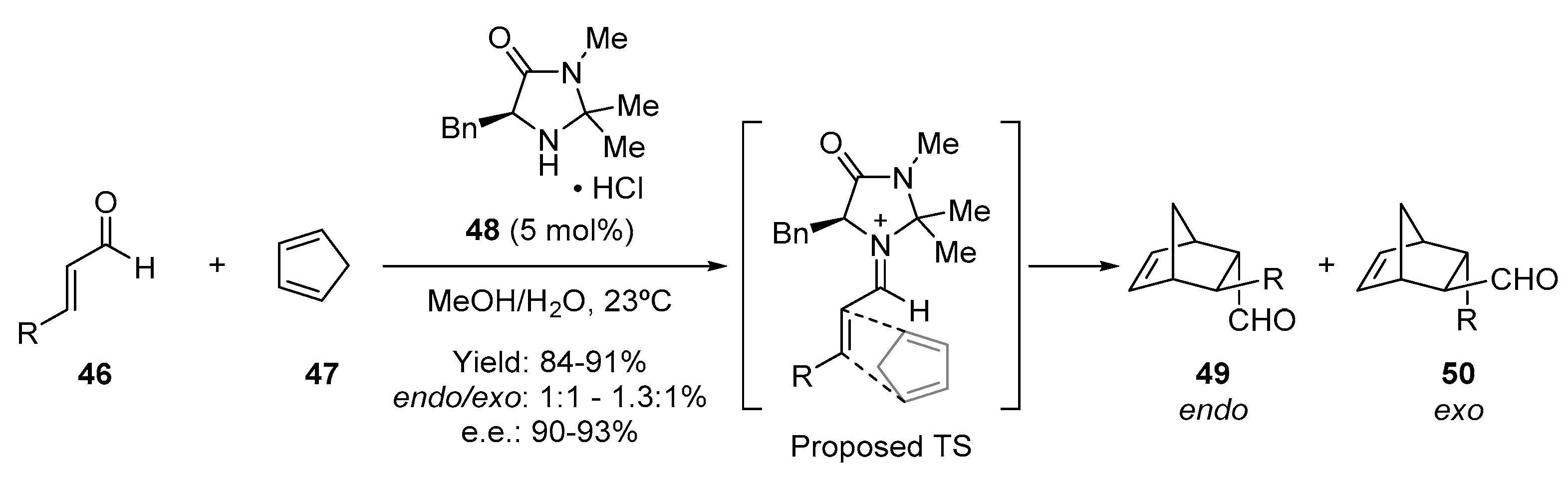
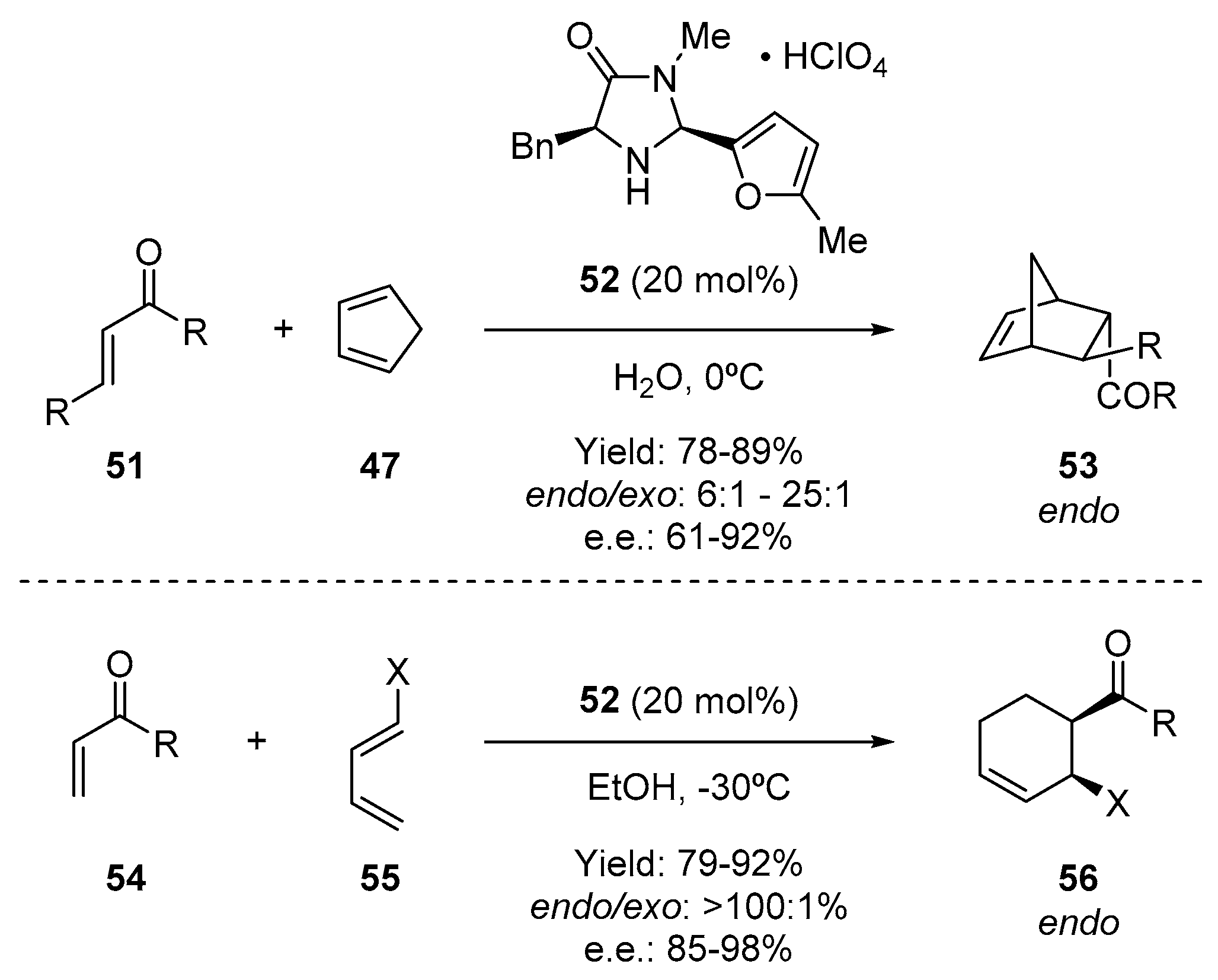
- Northrup, A.B.; MacMillan, D.W.C. The first general enantioselective catalytic Diels-Alder reaction with simple α,β-unsaturated ketones. J. Am. Chem. Soc. 2002, 124, 2458–2460. [Google Scholar] [CrossRef]
- Heravi, M.M.; Vavsari, V.F. Recent applications of intramolecular Diels—Alder reaction in total synthesis of natural products. RSC Adv. 2015, 5, 50890–50912. [Google Scholar] [CrossRef]
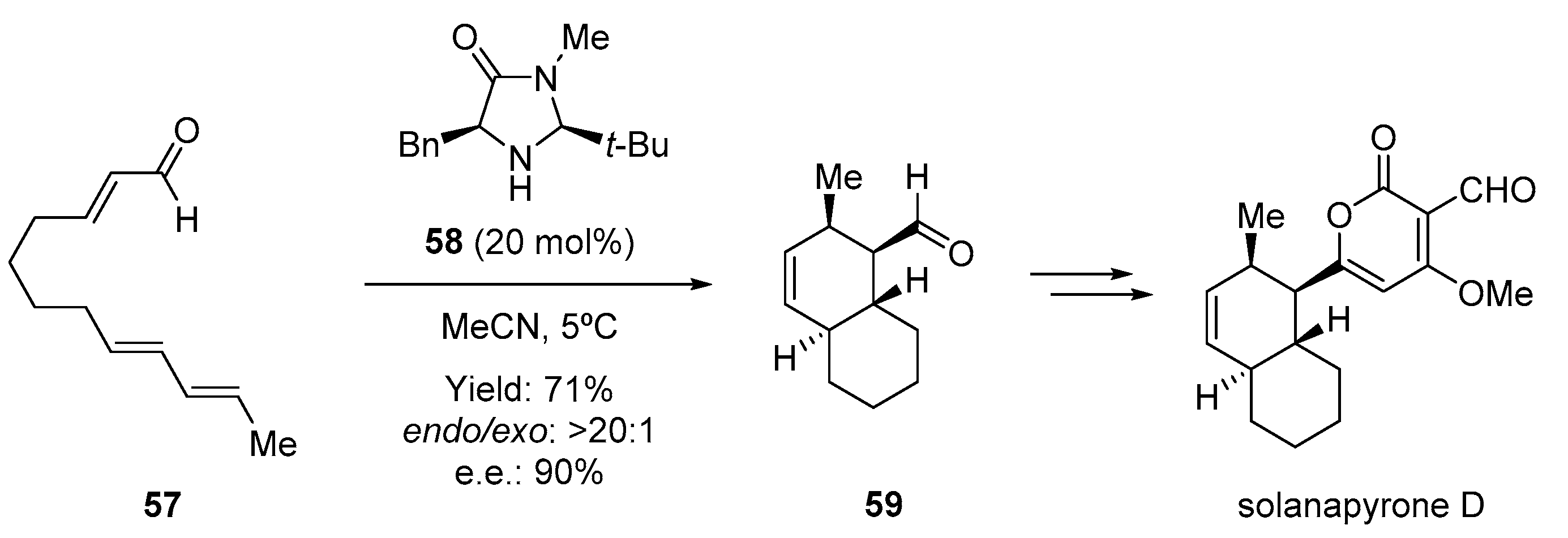
- Wilson, R.M.; Jen, W.S.; Macmillan, D.W.C. Enantioselective organocatalytic intramolecular Diels—Alder reactions. The asymmetric synthesis of solanapyrone D. J. Am. Chem. Soc. 2005, 127, 11616–11617. [Google Scholar] [CrossRef] [Green Version]
- Hagiwara, H.; Kobayashi, K.; Miya, S.; Hoshi, T.; Suzuki, T.; Ando, M.; Okamoto, T.; Kobayashi, M.; Yamamoto, I.; Ohtsubo, S.; et al. First total syntheses of the phytotoxins solanapyrones D and E via the domino Michael protocol. J. Org. Chem. 2002, 67, 5969–5976. [Google Scholar] [CrossRef]
- Jen, W.S.; Wiener, J.J.M.; Macmillan, D.W.C. New Strategies for Organic Catalysis: The First Enantioselective Organocatalytic 1,3-Dipolar Cycloaddition. J. Am. Chem. Soc. 2000, 122, 9874–9875. [Google Scholar] [CrossRef] [Green Version]
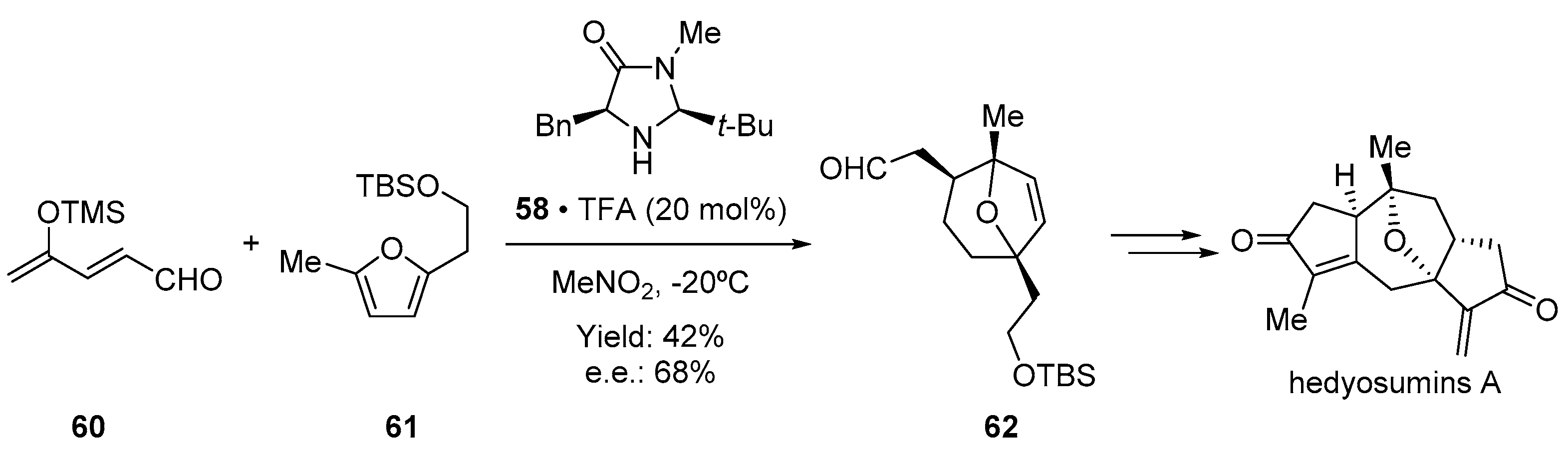
- Sun, W.-B.; Wang, X.; Sun, B.-F.; Zou, J.-P.; Lin, G.-Q. Catalytic Asymmetric Total Synthesis of Hedyosumins A, B, and C. Org. Lett. 2016, 18, 1219–1221. [Google Scholar] [CrossRef]
- Harmata, M.; Ghosh, S.K.; Hong, X.; Wacharasindhu, S.; Kirchhoefer, P. Asymmetric organocatalysis of 4 + 3 cycloaddition reactions. J. Am. Chem. Soc. 2003, 125, 2058–2059. [Google Scholar] [CrossRef] [PubMed]
- Erkkilä, A.; Majander, I.; Pihko, P.M. Iminium catalysis. Chem. Rev. 2007, 107, 5416–5470. [Google Scholar] [CrossRef] [PubMed]
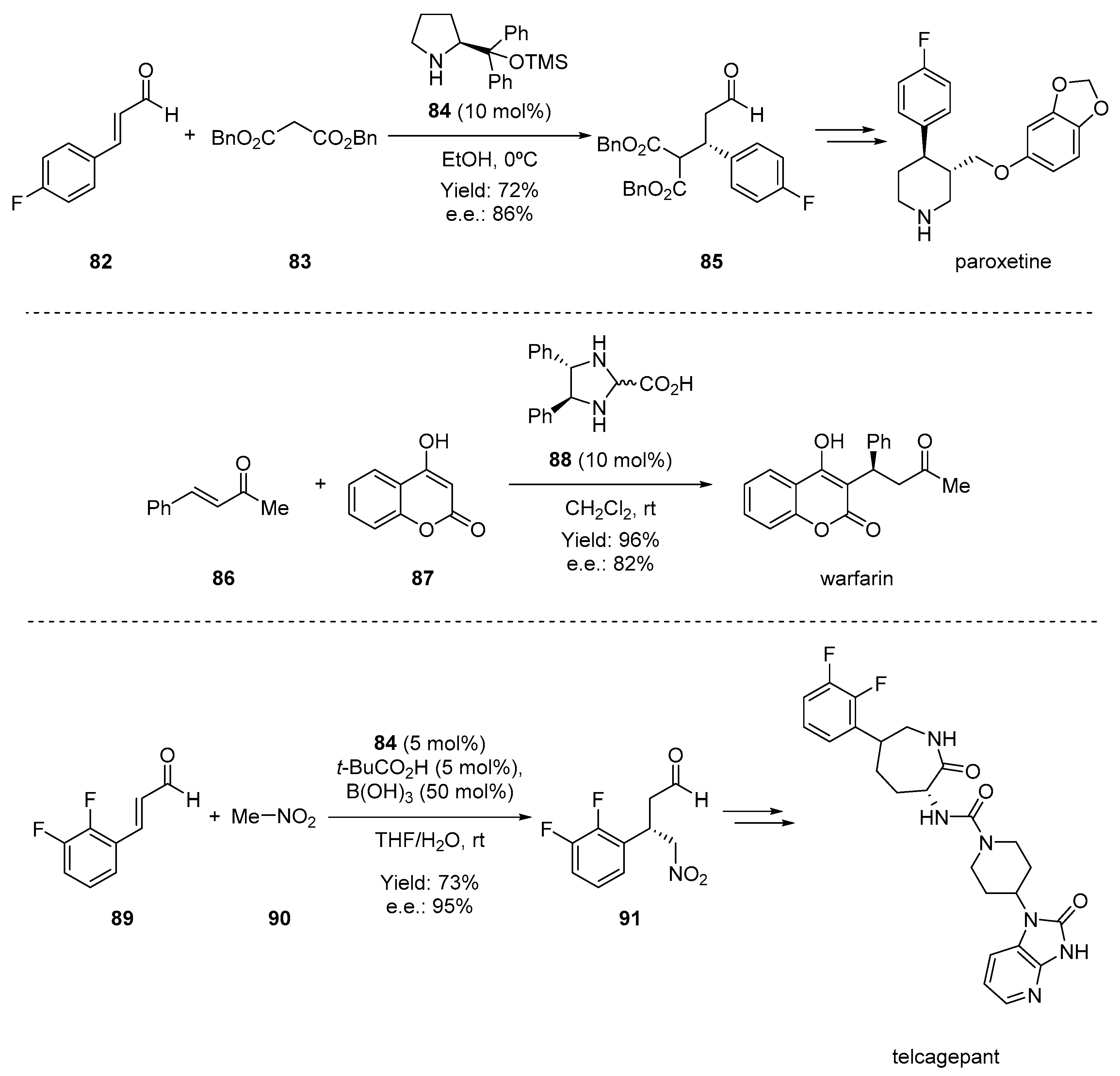
- Brandau, S.; Landa, A.; Franzén, J.; Marigo, M.; Jørgensen, K.A. Organocatalytic conjugate addition of malonates to α,β-unsaturated aldehydes: Asymmetric formal synthesis of (−)-paroxetine, chiral lactams, and lactones. Angew. Chem. Int. Ed. 2006, 45, 4305–4309. [Google Scholar] [CrossRef] [PubMed]
- Halland, N.; Hansen, T.; Jørgensen, K.A. Organocatalytic asymmetric Michael reaction of cyclic 1,3-dicarbonyl compounds and α,β-unsaturated ketones—A highly atom-economic catalytic one-step formation of optically active warfarin anticoagulant. Angew. Chem. Int. Ed. 2003, 42, 4955–4957. [Google Scholar] [CrossRef]
- Wittkowsky, A.K. Warfarin and other coumarin derivatives: Pharmacokinetics, pharmacodynamics, and drug interactions. Semin. Vasc. Med. 2003, 3, 221–230. [Google Scholar] [PubMed]
- Xu, F.; Zacuto, M.; Yoshikawa, N.; Desmond, R.; Hoerrner, S.; Itoh, T.; Journet, M.; Humphrey, G.R.; Cowden, C.; Strotman, N.; et al. Asymmetric synthesis of telcagepant, a CGRP receptor antagonist for the treatment of migraine. J. Org. Chem. 2010, 75, 7829–7841. [Google Scholar] [CrossRef] [PubMed]
- Paone, D.V.; Shaw, A.W.; Nguyen, D.N.; Burgey, C.S.; Deng, J.Z.; Kane, S.A.; Koblan, K.S.; Salvatore, C.A.; Mosser, S.D.; Johnston, V.K.; et al. Potent, orally bioavailable calcitonin gene-related peptide receptor antagonists for the treatment of migraine: Discovery of N-[(3R,6S)-6-(2,3-difluorophenyl)-2-oxo-1-(2,2,2-trifluoroethyl)azepan-3-yl]-4-(2-oxo-2,3-dihydro-1H-imidazo[4,5-b]pyridin- 1-yl)piperidine-1-carboxamide (MK-0974). J. Med. Chem. 2007, 50, 5564–5567. [Google Scholar]
- Starks, C.M. Phase-transfer catalysis. I. Heterogeneous reactions involving anion transfer by quaternary ammonium and phosphonium salts. J. Am. Chem. Soc. 1971, 93, 195–199. [Google Scholar] [CrossRef]
- Makosza, M.; Fedorynski, M. Phase Transfer Catalysis—Basic Principles, Mechanism and Specific Features. Curr. Catal. 2012, 1, 79–87. [Google Scholar] [CrossRef]
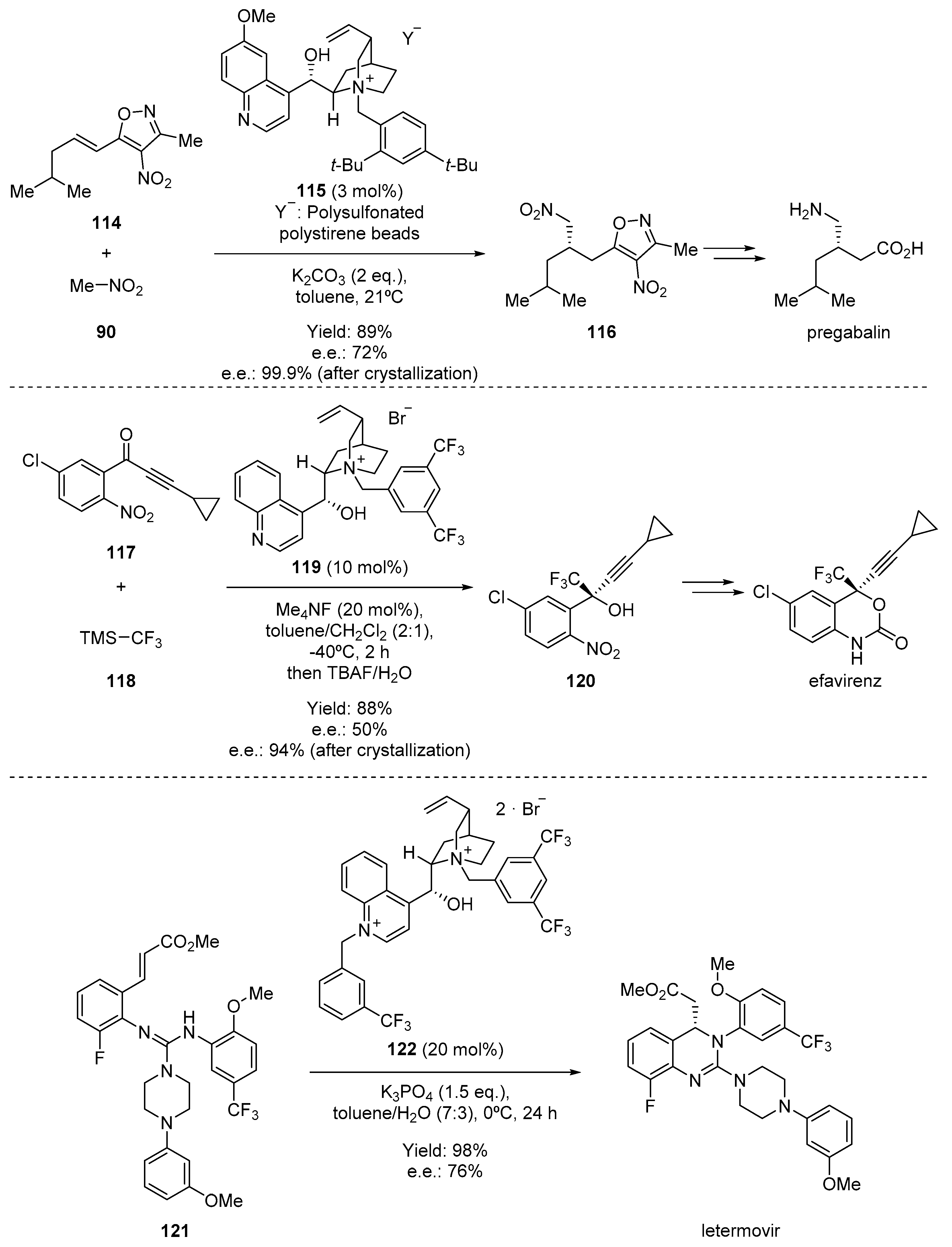
- Moccia, M.; Cortigiani, M.; Monasterolo, C.; Torri, F.; Del Fiandra, C.; Fuller, G.; Kelly, B.; Adamo, M.F.A. Development and Scale-up of an Organocatalytic Enantioselective Process to Manufacture (S)-Pregabalin. Org. Process. Res. Dev. 2015, 19, 1274–1281. [Google Scholar] [CrossRef]
- Kawai, H.; Kitayama, T.; Tokunaga, E.; Shibata, N. A New Synthetic Approach to Efavirenz through Enantioselective Trifluoromethylation by Using the Ruppert-Prakash Reagent. Eur. J. Org. Chem. 2011, 2011, 5959–5961. [Google Scholar] [CrossRef]
- Humphrey, G.R.; Dalby, S.M.; Andreani, T.; Xiang, B.; Luzung, M.R.; Song, Z.J.; Shevlin, M.; Christensen, M.; Belyk, K.M.; Tschaen, D.M. Asymmetric Synthesis of Letermovir Using a Novel Phase-Transfer-Catalyzed Aza-Michael Reaction. Org. Process. Res. Dev. 2016, 20, 1097–1103. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.S.; Jacobsen, E.N. Asymmetric catalysis by chiral hydrogen-bond donors. Angew. Chem. Int. Ed. 2006, 45, 1520–1543. [Google Scholar] [CrossRef] [PubMed]
- Corey, E.J.; Grogan, M.J. Enantioselective synthesis of α-amino nitriles from N-benzhydryl imines and HCN with a chiral bicyclic guanidine as catalyst. Org. Lett. 1999, 1, 157–160. [Google Scholar] [CrossRef]
- Vachal, P.; Jacobsen, E.N. Structure-based analysis and optimization of a highly enantioselective catalyst for the strecker reaction. J. Am. Chem. Soc. 2002, 124, 10012–10014. [Google Scholar] [CrossRef]
- Connon, S.J. Asymmetric catalysis with bifunctional cinchona alkaloid-based urea and thiourea organocatalysts. Chem. Commun. 2008, 2499–2510. [Google Scholar] [CrossRef]
- Yoon, T.P.; Jacobsen, E.N. Highly enantioselective thiourea-catalyzed nitro-Mannich reactions. Angew. Chem. Int. Ed. 2005, 44, 466–468. [Google Scholar] [CrossRef]
- Wenzel, A.G.; Jacobsen, E.N. Asymmetric catalytic Mannich reactions catalyzed by urea derivatives: Enantioselective synthesis of β-aryl-β-amino acids. J. Am. Chem. Soc. 2002, 124, 12964–12965. [Google Scholar] [CrossRef]
- Marcelli, T.; Hiemstra, H. Cinchona Alkaloids in Asymmetric Organocatalysis. Synthesis 2010, 8, 1229–1279. [Google Scholar] [CrossRef]
- Yu, X.; Wang, W. Hydrogen-bond-mediated asymmetric catalysis. Chem. Asian J. 2008, 3, 516–532. [Google Scholar] [CrossRef]
- Zhang, Z.; Bao, Z.; Xing, H. N,N’-Bis[3,5-bis(trifluoromethyl)phenyl]thiourea: A privileged motif for catalyst development. Org. Biomol. Chem. 2014, 12, 3151–3162. [Google Scholar] [CrossRef] [PubMed]
- Katharina, L.M.; Kira, H.; Dennis, G.; David, L.; Heike, H.; Sabine, G.; Peter, S.R. Hydrogen-Bonding Thiourea Organocatalysts: The Privileged 3,5-Bis(trifluoromethyl)phenyl Group. Eur. J. Org. Chem. 2012, 2012, 5919–5927. [Google Scholar]
- Akiyama, T.; Mori, K. Stronger Brønsted Acids: Recent Progress. Chem. Rev. 2015, 115, 9277–9306. [Google Scholar] [CrossRef] [PubMed]
- Entgelmeier, L.; Mancheño, O.G. Activation Modes in Asymmetric Anion-Binding Catalysis. Synthesis 2022, 54, 3907–3927. [Google Scholar]
- Connon, S.J. Chiral phosphoric acids: Powerful organocatalysts for asymmetric addition reactions to imines. Angew. Chem. Int. Ed. 2006, 45, 3909–3912. [Google Scholar] [CrossRef] [PubMed]
- Parmar, D.; Sugiono, E.; Raja, S.; Rueping, M. Complete field guide to asymmetric BINOL-phosphate derived Brønsted acid and metal catalysis: History and classification by mode of activation; Brønsted acidity, hydrogen bonding, ion pairing, and metal phosphates. Chem. Rev. 2014, 114, 9047–9153. [Google Scholar] [CrossRef]
- Melikian, M.; Gramüller, J.; Hioe, J.; Greindl, J.; Gschwind, R.M. Brønsted acid catalysis—the effect of 3,3’-substituents on the structural space and the stabilization of imine/phosphoric acid complexes. Chem. Sci. 2019, 10, 5226–5234. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, T.; Itoh, J.; Yokota, K.; Fuchibe, K. Enantioselective Mannich-Type Reaction Catalyzed by a Chiral Brønsted Acid. Angew. Chem. Int. Ed. 2004, 43, 1566–1568. [Google Scholar] [CrossRef]
- Uraguchi, D.; Terada, M. Chiral Brønsted acid-catalyzed direct Mannich reactions via electrophilic activation. J. Am. Chem. Soc. 2004, 126, 5356–5357. [Google Scholar] [CrossRef]
- Xu, F.; Corley, E.; Zacuto, M.; Conlon, D.A.; Pipik, B.; Humphrey, G.; Murry, J.; Tschaen, D. Asymmetric synthesis of a potent, aminopiperidine-fused imidazopyridine dipeptidyl peptidase IV inhibitor. J. Org. Chem. 2010, 75, 1343–1353. [Google Scholar] [CrossRef]
- Jakubec, P.; Cockfield, D.M.; Dixon, D.J. Total synthesis of (−)-nakadomarin A. J. Am. Chem. Soc. 2009, 131, 16632–16633. [Google Scholar] [CrossRef] [PubMed]
- Xiaojun, H.; Scott, B.; Charles, D.; Shu-Hai, Z. Pilot-Plant Preparation of 3,4-Dihydropyridin-2-one Derivatives, the Core Structures of P2 × 7 Receptor Antagonists. Organ. Process Res. Dev. 2010, 14, 612–616. [Google Scholar]























Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reyes, E.; Prieto, L.; Milelli, A. Asymmetric Organocatalysis: A Survival Guide to Medicinal Chemists. Molecules 2023, 28, 271. https://doi.org/10.3390/molecules28010271
Reyes E, Prieto L, Milelli A. Asymmetric Organocatalysis: A Survival Guide to Medicinal Chemists. Molecules. 2023; 28(1):271. https://doi.org/10.3390/molecules28010271
Chicago/Turabian StyleReyes, Efraim, Liher Prieto, and Andrea Milelli. 2023. "Asymmetric Organocatalysis: A Survival Guide to Medicinal Chemists" Molecules 28, no. 1: 271. https://doi.org/10.3390/molecules28010271
APA StyleReyes, E., Prieto, L., & Milelli, A. (2023). Asymmetric Organocatalysis: A Survival Guide to Medicinal Chemists. Molecules, 28(1), 271. https://doi.org/10.3390/molecules28010271