

New Class of Benzodiazepinone Derivatives as Pro-Death Agents Targeting BIR Domains in Cancer Cells

 , , , ,
, , , ,  and
and
Abstract
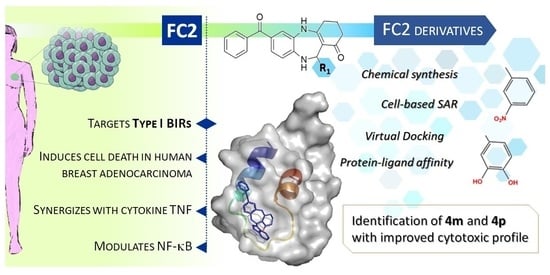
:
1. Introduction
2. Results
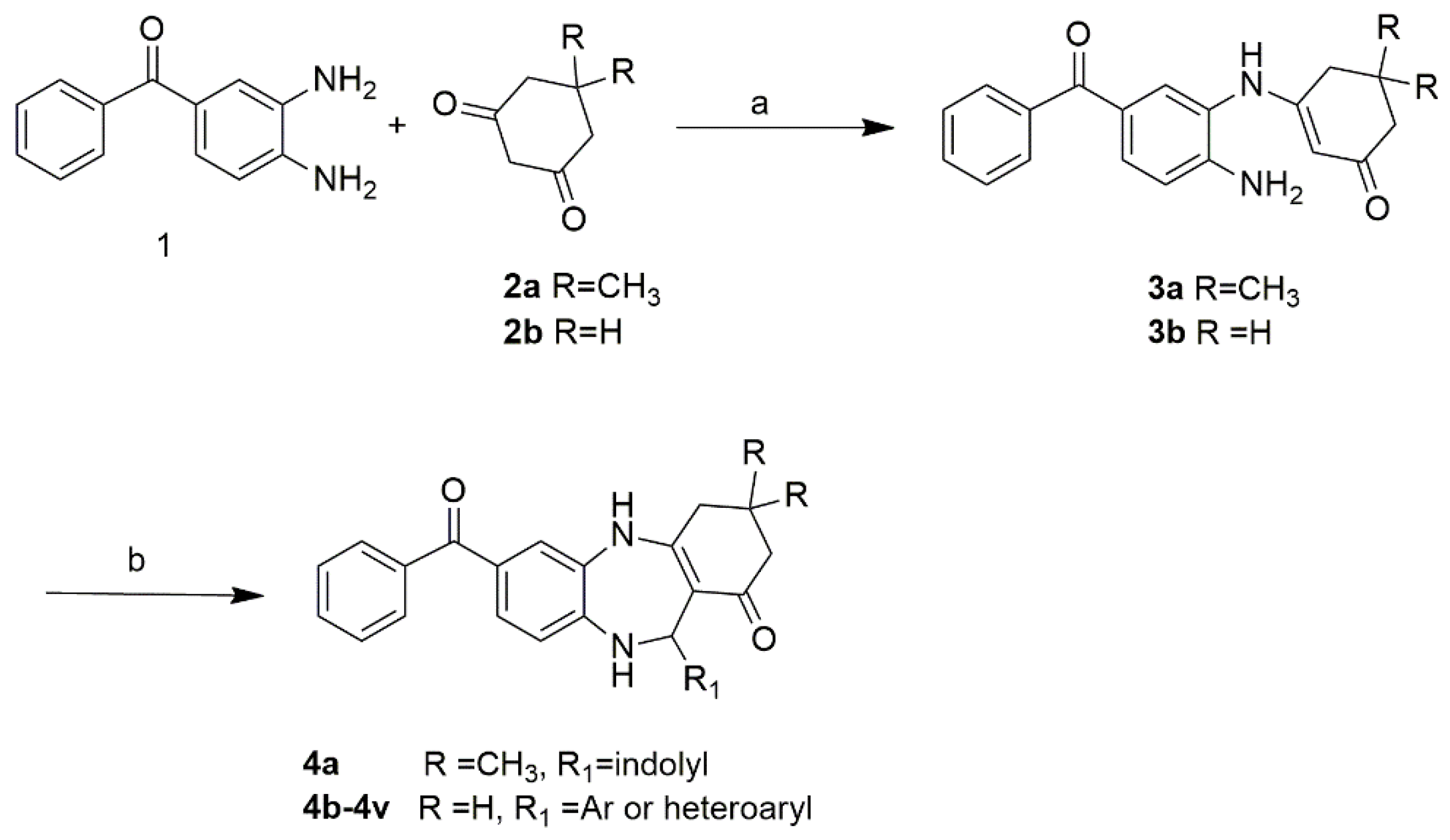
2.1. Chemistry
2.2. SAR of FC2 Derivatives on MDA-MB-231 Human Breast Adenocarcinoma
2.2.1. Compound 4d As a Novel Base for the Design of New Putative NF-κB Modulators
2.2.2. Identification of the Best Mono-Substitutions of R1
2.2.3. Di- and Tri-substituted Derivatives
2.3. Cytotoxic Effect of the Derivatives in Combination with TNF
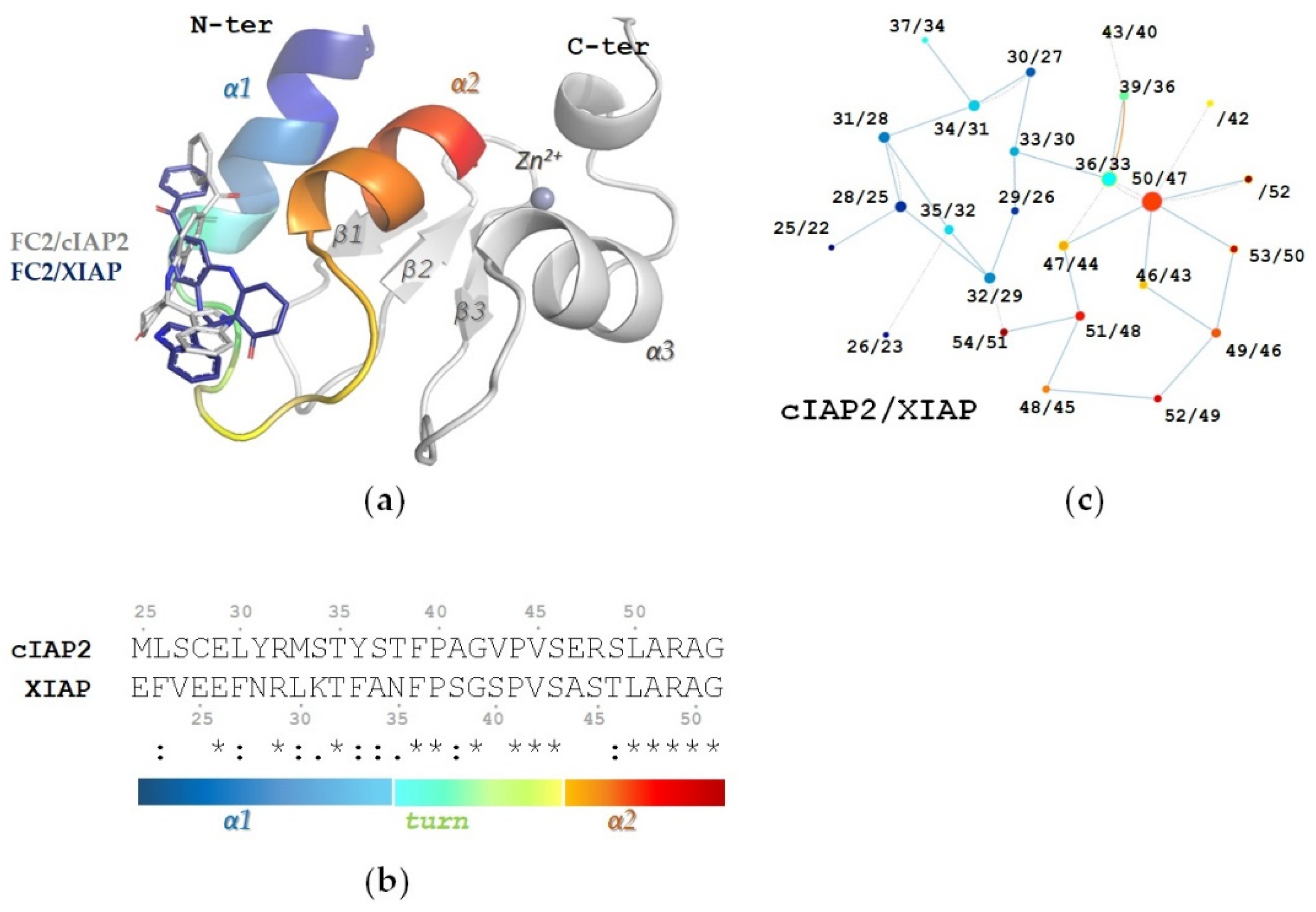
2.4. In Silico Characterization of FC2 Derivatives Binding to Type I BIRs
2.4.1. The R1 Modification Is Relevant to the Positioning of FC2 Derivatives in the Structural Hotspot
2.4.2. Cytotoxic FC2 Derivatives Retain FC2 Pose and Interaction Network in the BIR1 Structural Hotspot
2.4.3. Electron Withdrawing Groups in the Meta Position of R1 Improve the Interaction Network
2.5. Effect of FC2 Derivatives on Type I BIRs of cIAP2 and XIAP
2.5.1. Thermal Stability
2.5.2. Measurement of FC2 Derivatives’ Affinity for Type I BIRs
3. Discussion
4. Materials and Methods
4.1. Chemistry
4.1.1. Experimental Instrumentation
4.1.2. Synthesis of FC2 and FC2 Derivatives (4a-e)
7-benzoyl-11-(1H-indol-3-yl)-2,3,4,5,10,11-hexahydro-1H-dibenzo[b,e][1,4]diazepin-1-one (FC2)
7-benzoyl-11-(1H-indol-3-yl)-3,3-dimethyl-2,3,4,5,10,11-hexahydro-1H-dibenzo[b,e][1,4]diazepin-1-one (4a)
7-benzoyl-11-(thiophen-2-yl)-2,3,4,5,10,11-hexahydro-1H-dibenzo[b,e][1,4]diazepin-1-one (4b)
7-benzoyl-11-(5-methylfuran-2-yl)-2,3,4,5,10,11-hexahydro-1H-dibenzo[b,e][1,4]diazepin-1-one (4c)
7-benzoyl-11-phenyl-2,3,4,5,10,11-hexahydro-1H-dibenzo[b,e][1,4]diazepin-1-one (4d)
7-benzoyl-11-(p-tolyl)-2,3,4,5,10,11-hexahydro-1H-dibenzo[b,e][1,4]diazepin-1-one (4e)
4.2. Cell Culturing and Cell Viability Assays
4.3. Virtual Docking
4.4. Cloning, Expression and Purification of the BIR1 Domains of cIAP2 and XIAP
4.5. Thermal Stability Assays
4.6. Binding Assays
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Greenblatt, D.J.; Shader, R.I.; Abernethy, D.R. Drug Therapy. Current Status of Benzodiazepines. N. Engl. J. Med. 1983, 309, 354–358. [Google Scholar]
- Mandelli, M.; Tognoni, G.; Garattini, S. Clinical Pharmacokinetics of Diazepam. Clin. Pharmacokinet. 1978, 3, 72–91. [Google Scholar] [CrossRef]
- Henderson, E.A.; Alber, D.G.; Baxter, R.C.; Bithell, S.K.; Budworth, J.; Carter, M.C.; Chubb, A.; Cockerill, G.S.; Dowdell, V.C.L.; Fraser, I.J.; et al. 1,4-Benzodiazepines as Inhibitors of Respiratory Syncytial Virus. The Identification of a Clinical Candidate. J. Med. Chem. 2007, 50, 1685–1692. [Google Scholar] [CrossRef] [PubMed]
- Micale, N.; Colleoni, S.; Postorino, G.; Pellicanò, A.; Zappalà, M.; Lazzaro, J.; Diana, V.; Cagnotto, A.; Mennini, T.; Grasso, S. Structure-Activity Study of 2,3-Benzodiazepin-4-Ones Noncompetitive AMPAR Antagonists: Identification of the 1-(4-Amino-3-Methylphenyl)-3,5-Dihydro-7,8-Ethylenedioxy-4H-2,3-Benzodiazepin-4-One as Neuroprotective Agent. Bioorg. Med. Chem. 2008, 16, 2200–2211. [Google Scholar] [CrossRef]
- Leonard, K.; Marugan, J.J.; Raboisson, P.; Calvo, R.; Gushue, J.M.; Koblish, H.K.; Lattanze, J.; Zhao, S.; Cummings, M.D.; Player, M.R.; et al. Novel 1,4-Benzodiazepine-2,5-Diones as Hdm2 Antagonists with Improved Cellular Activity. Bioorg. Med. Chem. Lett. 2006, 16, 3463–3468. [Google Scholar] [CrossRef] [PubMed]
- Gourdeau, H.; McAlpine, J.B.; Ranger, M.; Simard, B.; Berger, F.; Beaudry, F.; Farnet, C.M.; Falardeau, P. Identification, Characterization and Potent Antitumor Activity of ECO-4601, a Novel Peripheral Benzodiazepine Receptor Ligand. Cancer Chemother. Pharmacol. 2008, 61, 911–921. [Google Scholar] [CrossRef]
- Mason, W.P.; Belanger, K.; Nicholas, G.; Vallières, I.; Mathieu, D.; Kavan, P.; Desjardins, A.; Omuro, A.; Reymond, D. A Phase II Study of the Ras-MAPK Signaling Pathway Inhibitor TLN-4601 in Patients with Glioblastoma at First Progression. J. Neurooncol. 2012, 107, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Casellas, P.; Galiegue, S.; Basile, A.S. Peripheral Benzodiazepine Receptors and Mitochondrial Function. Neurochem. Int. 2002, 40, 475–486. [Google Scholar] [CrossRef]
- Han, Z.; Slack, R.S.; Li, W.; Papadopoulos, V. Expression of Peripheral Benzodiazepine Receptor (PBR) in Human Tumors: Relationship to Breast, Colorectal, and Prostate Tumor Progression. J. Recept. Signal Transduct. Res. 2003, 23, 225–238. [Google Scholar] [CrossRef]
- Castedo, M.; Perfettini, J.-L.; Kroemer, G. Mitochondrial Apoptosis and the Peripheral Benzodiazepine Receptor: A Novel Target for Viral and Pharmacological Manipulation. J. Exp. Med. 2002, 196, 1121–1125. [Google Scholar] [CrossRef] [Green Version]
- Decaudin, D.; Castedo, M.; Nemati, F.; Beurdeley-Thomas, A.; De Pinieux, G.; Caron, A.; Pouillart, P.; Wijdenes, J.; Rouillard, D.; Kroemer, G.; et al. Ligands Reverse Apoptosis Resistance of Cancer Cells in Vitro and in Vivo. Cancer Res. 2002, 62, 1388–1393. [Google Scholar]
- Donnell, A.F.; Michoud, C.; Rupert, K.C.; Han, X.; Aguilar, D.; Frank, K.B.; Fretland, A.J.; Gao, L.; Goggin, B.; Hogg, J.H.; et al. Benzazepinones and Benzoxazepinones as Antagonists of Inhibitor of Apoptosis Proteins (IAPs) Selective for the Second Baculovirus IAP Repeat (BIR2) Domain. J. Med. Chem. 2013, 56, 7772–7787. [Google Scholar] [CrossRef]
- Kester, R.F.; Donnell, A.F.; Lou, Y.; Remiszewski, S.W.; Lombardo, L.J.; Chen, S.; Le, N.T.; Lo, J.; Moliterni, J.A.; Han, X.; et al. Optimization of Benzodiazepinones as Selective Inhibitors of the X-Linked Inhibitor of Apoptosis Protein (XIAP) Second Baculovirus IAP Repeat (BIR2) Domain. J. Med. Chem. 2013, 56, 7788–7803. [Google Scholar] [CrossRef]
- Cossu, F.; Camelliti, S.; Lecis, D.; Sorrentino, L.; Majorini, M.T.; Milani, M.; Mastrangelo, E. Structure-Based Identification of a New IAP-Targeting Compound That Induces Cancer Cell Death Inducing NF-κB Pathway. Comput. Struct. Biotechnol. J. 2021, 19, 6366–6374. [Google Scholar] [CrossRef]
- Zheng, C.; Kabaleeswaran, V.; Wang, Y.; Cheng, G.; Wu, H. Crystal Structures of the TRAF2: cIAP2 and the TRAF1: TRAF2: cIAP2 Complexes: Affinity, Specificity, and Regulation. Mol. Cell 2010, 38, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Lin, S.-C.; Huang, Y.; Kang, Y.J.; Rich, R.; Lo, Y.-C.; Myszka, D.; Han, J.; Wu, H. XIAP Induces NF-kappaB Activation via the BIR1/TAB1 Interaction and BIR1 Dimerization. Mol. Cell 2007, 26, 689–702. [Google Scholar] [CrossRef] [Green Version]
- Rathore, R.; McCallum, J.E.; Varghese, E.; Florea, A.-M.; Büsselberg, D. Overcoming Chemotherapy Drug Resistance by Targeting Inhibitors of Apoptosis Proteins (IAPs). Apoptosis 2017, 22, 898–919. [Google Scholar] [CrossRef] [Green Version]
- Petersen, S.L.; Peyton, M.; Minna, J.D.; Wang, X. Overcoming Cancer Cell Resistance to Smac Mimetic Induced Apoptosis by Modulating cIAP-2 Expression. Proc. Natl. Acad. Sci. USA 2010, 107, 11936–11941. [Google Scholar] [CrossRef] [Green Version]
- Mehrazar, M.; Hassankalhori, M.; Toolabi, M.; Goli, F.; Moghimi, S.; Nadri, H.; Bukhari, S.N.A.; Firoozpour, L.; Foroumadi, A. Design and Synthesis of Benzodiazepine-1,2,3-Triazole Hybrid Derivatives as Selective Butyrylcholinesterase Inhibitors. Mol. Divers. 2020, 24, 997–1013. [Google Scholar] [CrossRef]
- Adib, M.; Zainali, M.; Kim, I. An Efficient Three-Component Synthesis of benzimidazo[1,2-A]-Quinoline-6-Carbonitriles. Synlett 2016, 27, 1844–1847. [Google Scholar] [CrossRef]
- Strakov, A.Y.; Petrova, M.V.; Tonkikh, N.N.; Gurkovskii, A.I.; Popelis, Y.; Kreishman, G.P.; Belyakov, S.V. Dibenzodiazepines in Reactions of 2-Acetyl-Dimedone with 3,4-Diaminobenzophenone. Chem. Heterocycl. Compd. 1997, 33, 321–332. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, L.; DeLano, W. PyMOL. 2020. Available online: http://www.pymol.org/pymo (accessed on 28 December 2022).
- Piovesan, D.; Minervini, G.; Tosatto, S.C.E. The RING 2.0 Web Server for High Quality Residue Interaction Networks. Nucleic Acids Res. 2016, 44, W367–W374. [Google Scholar] [CrossRef]
- Cossu, F.; Milani, M.; Mastrangelo, E.; Lecis, D. Targeting the BIR Domains of Inhibitor of Apoptosis (IAP) Proteins in Cancer Treatment. Comput. Struct. Biotechnol. J. 2019, 17, 142–150. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Sanner, M.F. Python: A Programming Language for Software Integration and Development. J. Mol. Graph. Mod. 1999, 17, 57–61. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R * | R1 * | EC50 [μM] ** | EC50 (+TNF) [μM] ** | ||
|---|---|---|---|---|---|
| FC2 | H | indole | 11.38 ± 2.08 | 9.08 ± 1.85 | |
| 4a | CH3 | indole | 42.76 ± 10.10 | 16.91 ± 7.14 | |
| 4b | H | thiophene | 21.90 ± 8.17 | 9.20 ± 5.75 | |
| 4c | H | 2-methylfuran | 65.11 ± 2.43 | 45.29 ± 3.75 | |
| 4d | H | phenyl | 19.57 ± 4.23 | 69.25 ± 8.10 | |
| 4e | H | phenyl - | CH3 (para) | 38.87 ± 7.55 | 47.9 ± 9.70 |
| 4f | H | phenyl - | N(CH3)2 (para) | 39.11 ± 11.10 | 24.21 ± 10.40 |
| 4g | H | phenyl - | OCH3 (para) | 67.23 ± 7.60 | 36.31 ± 10.45 |
| 4h | H | phenyl - | CF3 (para) | 22.32 ± 8.67 | 48.77 ± 5.72 |
| 4i | H | phenyl - | Cl (para) | 25.89 ± 7.03 | 22.15 ± 6.67 |
| 4j | H | phenyl - | OH (para) | 48.18 ± 15.10 | 59.74 ± 14.8 |
| 4k | H | phenyl - | OCH3 (meta) | 10.26 ± 4.63 | 12.99 ± 4.15 |
| 4l | H | phenyl - | OH (meta) | 18.58 ± 2.56 | 16.07 ± 3.70 |
| 4m | H | phenyl - | NO2 (meta) | 5.54 ± 1.03 | 3.82 ± 1.41 |
| 4n | H | phenyl - | NH2 (meta) | NA *** | NA |
| 4o | H | phenyl - | OCH3, OCH3 (para, meta) | 13.47 ± 4.67 | 15.44 ± 6.86 |
| 4p | H | phenyl - | OH, OH (para, meta) | 6.92 ± 2.43 | 9.50 ± 4.73 |
| 4q | H | phenyl - | 2,2-difluorobenzo[d][1,3]dioxole | 14.68 ± 4.98 | 14.04 ± 2.40 |
| 4r | H | phenyl - | OH, OCH3 (para, meta) | 7.49 ± 1.65 | 12.85 ± 1.90 |
| 4s | H | phenyl - | OCH3, OH (para, meta) | 15.25 ± 3.63 | 14.60 ± 1.74 |
| 4t | H | phenyl - | OH, OCH3 (ortho, meta) | NA | NA |
| 4u | H | phenyl - | OH, NO2 (para, meta) | 17.03 ± 5.40 | 9.53 ± 3.61 |
| 4v | H | phenyl - | OCH3, OH, NO2 (meta, para, meta) | 31.45 ± 11.4 | 26.81 ± 12.5 |
| cIAP2-BIR1 | XIAP-BIR1 | |||||
|---|---|---|---|---|---|---|
| Ki (μM) * | ΔTM (°C) ** | Kd (μM) *** | Ki (μM) * | ΔTM (°C) ** | Kd (μM) *** | |
| FC2 | 1.00 | −2.10 | 0.50 ± 0.20 | 1.00 | −2.90 | 5.20 ± 1.00 |
| 4a | 0.63 | 0.35 | 1.30 ± 0.70 | 1.20 | 0.30 | 6.80 ± 1.30 |
| 4b | 9.09 | −18.69 | 46.30 ± 24.80 | 1.36 | −9.66 | 30.00 ± 7.47 |
| 4c | 5.17 | −21.05 | >50 | 0.89 | −12.28 | >50 |
| 4d | 2.20 | 0.45 | 21.90 ± 6.20 | 2.34 | −0.30 | 4.40 ± 2.20 |
| 4e | 4.68 | −1.35 | 27.60 ± 7.30 | 2.23 | 0.72 | >50 |
| 4f | 5.61 | −17.84 | 38.70 ± 12.20 | 2.14 | 1.31 | 27.55 3.16 |
| 4g | 3.10 | 0.75 | 5.40 ± 1.50 | 4.83 | −0.50 | 18.00 ± 5.40 |
| 4h | 5.29 | −2.00 | 43.40 ± 13.90 | 2.23 | −13.41 | 14.90 ± 3.80 |
| 4i | 2.80 | −1.70 | >50 | 1.00 | 0.15 | >50 |
| 4j | 2.10 | −18.80 | 27.55 ± 3.16 | 2.04 | −8.90 | 7.26 ± 0.98 |
| 4k | 3.26 | −0.20 | >50 | 2.11 | 0.15 | >50 |
| 4l | 2.80 | −2.60 | 38.58 ± 3.56 | 3.70 | −12.50 | 22.00 ± 1.18 |
| 4m | 1.20 | 0.35 | 20.50 ± 4.00 | 0.20 | 0.25 | 4.30 ± 1.80 |
| 4n | 2.30 | −2.10 | 26.53 ± 1.07 | 1.60 | −16.80 | 38.06 ± 1.68 |
| 4o | 5.20 | −2.55 | 12.00 ± 2.40 | 3.20 | −0.50 | 7.50 ± 2.90 |
| 4p | 7.00 | −0.60 | 26.90 ± 4.30 | 3.32 | −1.95 | 15.90 ± 5.20 |
| 4q | 3.00 | −0.60 | 16.40 ± 2.90 | 1.10 | 0.35 | 41.80 ± 11.70 |
| 4r | 7.00 | −2.10 | 46.05 ± 8.33 | 1.30 | −1.40 | 33.07 ± 1.40 |
| 4s | 1.70 | −2.50 | >50 | 1.50 | −12.40 | >50 |
| 4t | 6.70 | −6.50 | >50 | 1.03 | −11.40 | >50 |
| 4u | 0.94 | −14.80 | >50 | 1.77 | −7.80 | 37.30 ± 4.45 |
| 4v | 5.50 | −13.1 | 38.04 ± 2.78 | 5.60 | −6.20 | 46.41 ± 5.31 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiore, M.; Mosconi, M.; Bonì, F.; Parodi, A.; Salis, A.; Tasso, B.; Mastrangelo, E.; Millo, E.; Cossu, F. New Class of Benzodiazepinone Derivatives as Pro-Death Agents Targeting BIR Domains in Cancer Cells. Molecules 2023, 28, 446. https://doi.org/10.3390/molecules28010446
Fiore M, Mosconi M, Bonì F, Parodi A, Salis A, Tasso B, Mastrangelo E, Millo E, Cossu F. New Class of Benzodiazepinone Derivatives as Pro-Death Agents Targeting BIR Domains in Cancer Cells. Molecules. 2023; 28(1):446. https://doi.org/10.3390/molecules28010446
Chicago/Turabian StyleFiore, Michele, Michele Mosconi, Francesco Bonì, Alice Parodi, Annalisa Salis, Bruno Tasso, Eloise Mastrangelo, Enrico Millo, and Federica Cossu. 2023. "New Class of Benzodiazepinone Derivatives as Pro-Death Agents Targeting BIR Domains in Cancer Cells" Molecules 28, no. 1: 446. https://doi.org/10.3390/molecules28010446
APA StyleFiore, M., Mosconi, M., Bonì, F., Parodi, A., Salis, A., Tasso, B., Mastrangelo, E., Millo, E., & Cossu, F. (2023). New Class of Benzodiazepinone Derivatives as Pro-Death Agents Targeting BIR Domains in Cancer Cells. Molecules, 28(1), 446. https://doi.org/10.3390/molecules28010446