

Microwave-Assisted Synthesis of Aminophosphonic Derivatives and Their Antifungal Evaluation against Lomentospora prolificans

 , ,
, ,  , , and
, , and
Abstract
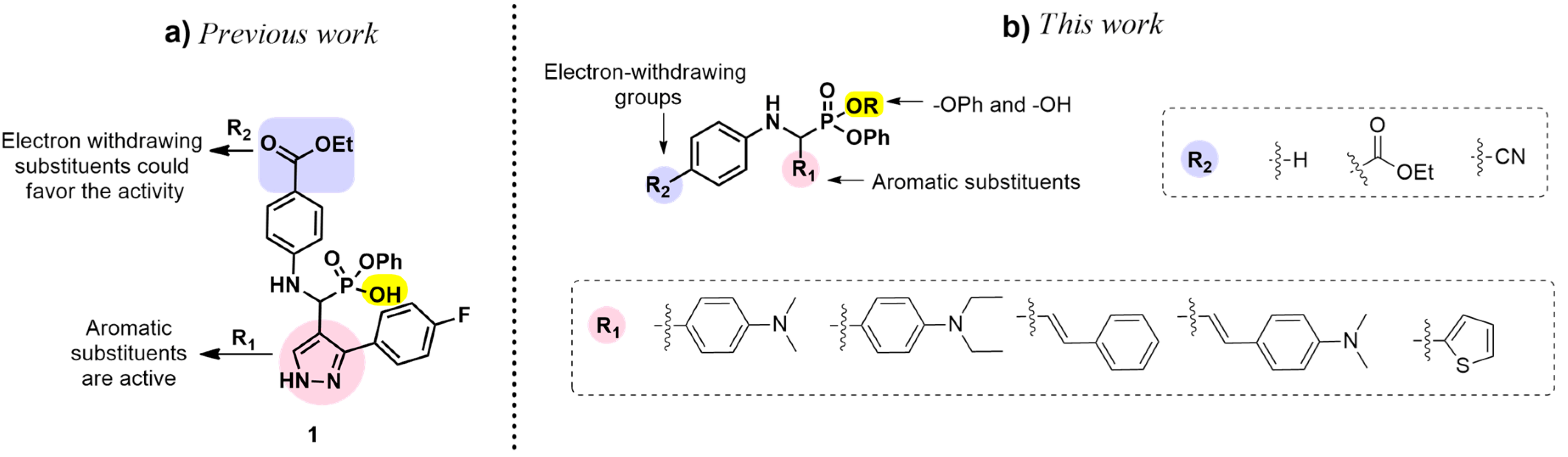
:1. Introduction
2. Results and Discussion
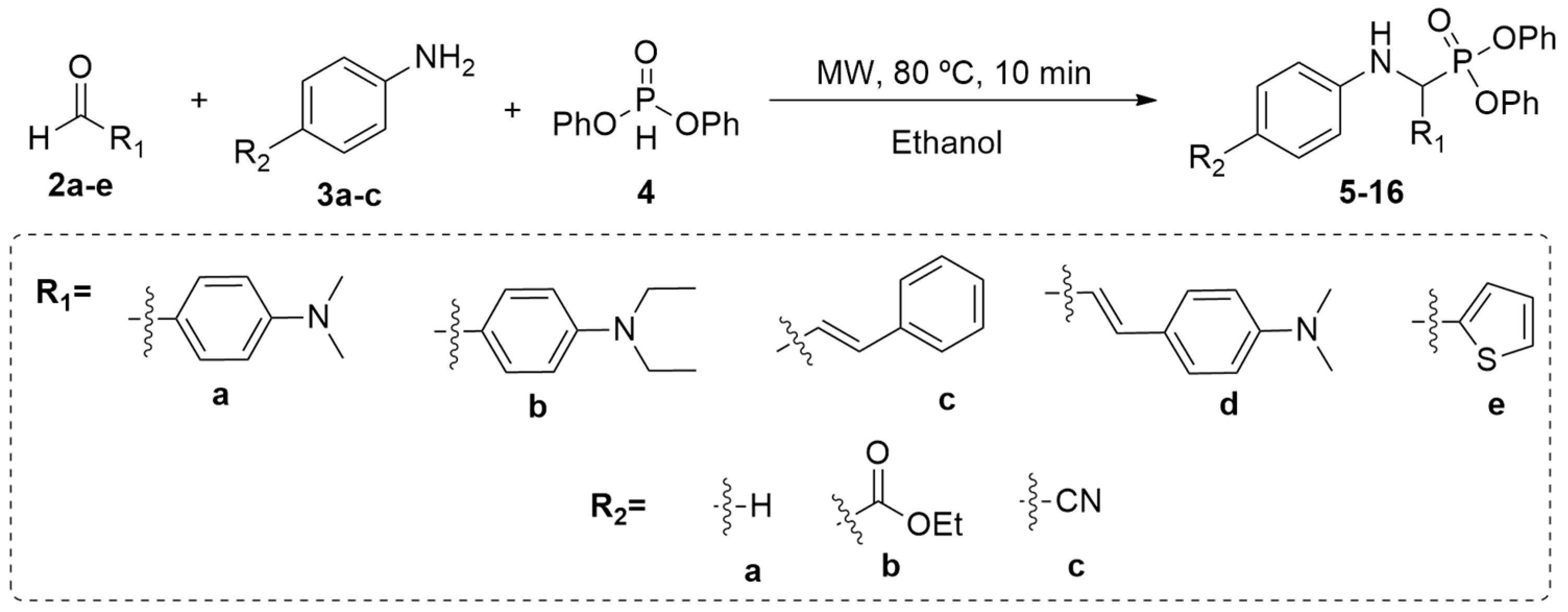
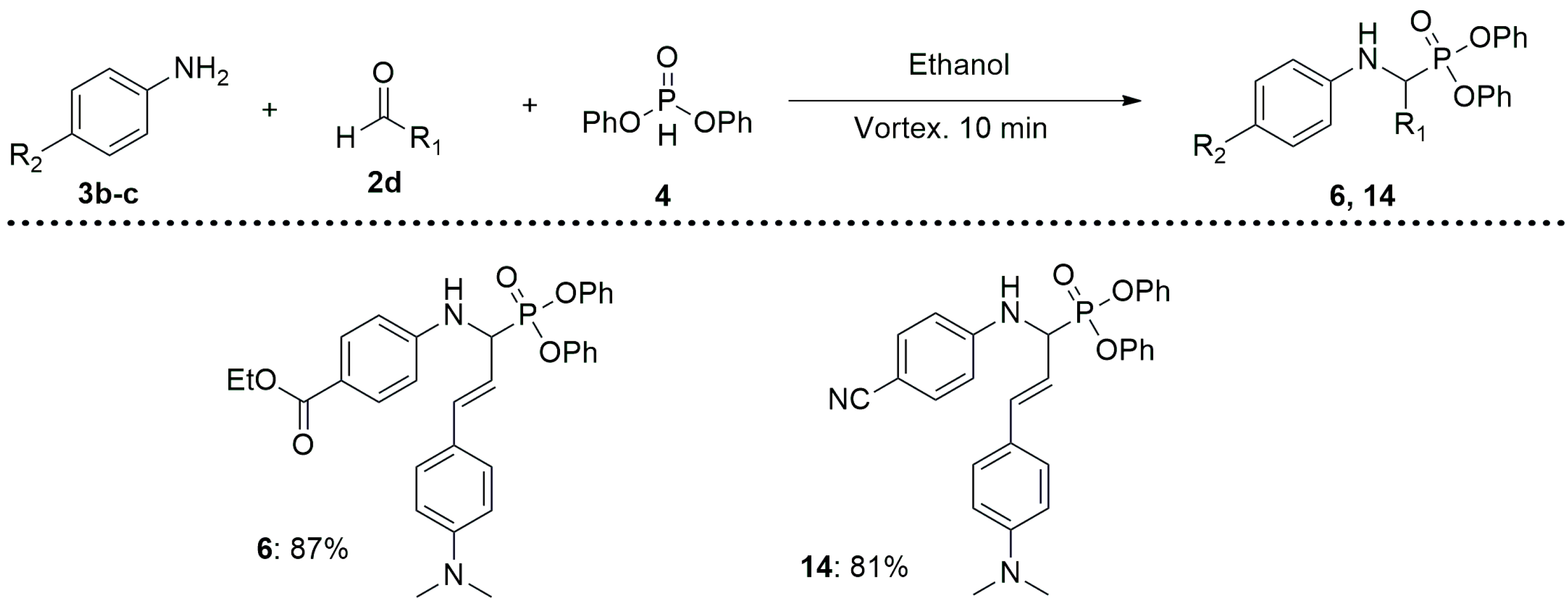
2.1. Chemistry
2.2. In Vitro Assays
2.3. Antifungal Activity Evaluation (M38-A2 Protocol)
2.4. Molecular Docking
2.5. Cytotoxicity
3. Materials and Methods
3.1. Synthesis of α-Aminophosphonates 5–16
- (E)-ethyl 4-((1-(diphenoxyphosphoryl)-3-phenylallyl)amino)benzoate (5)
- (E)-ethyl 4-((3-(4-(dimethylamino)phenyl)-1-(diphenoxyphosphoryl)allyl)amino) benzoate (6)
- Ethyl 4-(((4-(dimethylamino)phenyl)(diphenoxyphosphoryl)methyl)amino)benzoate (7)
- Ethyl 4-(((4-(diethylamino)phenyl)(diphenoxyphosphoryl)methyl)amino)benzoate (8)
- Ethyl 4-(((diphenoxyphosphoryl)(thiophen-2-yl)methyl)amino)benzoate (9)
- (E)-diphenyl (3-phenyl-1-(phenylamino)allyl)phosphonate (10)
- Diphenyl ((4-(dimethylamino)phenyl)(phenylamino)methyl)phosphonate (11)
- Diphenyl ((phenylamino)(thiophen-2-yl)methyl)phosphonate (12)
- (E)-diphenyl (1-((4-cyanophenyl)amino)-3-phenyl allyl)phosphonate (13)
- (E)-diphenyl(1-((4-cyanophenyl)amino)-3-(4-(dimethylamino)phenyl)allyl)phosphonate (14)
- Diphenyl (((4-cyanophenyl)amino)(4-(dimethylamino)phenyl)methyl)phosphonate (15)
- Diphenyl (((4-cyanophenyl)amino)(4-(diethylamino)phenyl)methyl)phosphonate (16)
3.2. Synthesis of α-Aminophosphonic Acids 17–28
- (E)-ethyl 4-((1-(hydroxy(phenoxy)phosphoryl)-3-phenylallyl)amino)benzoate (17)
- (E)-ethyl 4-((3-(4-(dimethylamino)phenyl)-1-(hydroxy(phenoxy)phosphoryl)allyl) amino)benzoate (18)
- Ethyl 4-(((4-(dimethylamino)phenyl)(hydroxy(phenoxy)phosphoryl)methyl)amino)benzoate (19)
- Ethyl 4-(((4-(diethylamino)phenyl)(hydroxy(phenoxy)phosphoryl)methyl)amino) benzoate (20)
- Ethyl 4-(((hydroxy(phenoxy)phosphoryl)(thiophen-2-yl)methyl)amino)benzoate (21)
- (E)-phenyl hydrogen (3-phenyl-1-(phenylamino)allyl)phosphonate (22)
- Phenyl hydrogen ((phenylamino)(thiophen-2-yl)methyl)phosphonate (23)
- (E)-phenyl hydrogen (1-((4-cyanophenyl)amino)-3-phenylallyl)phosphonate (24)
- (E)-phenyl hydrogen (1-((4-cyanophenyl)amino)-3-(4-(dimethylamino)phenyl)allyl) phosphonate (25)
- Phenyl hydrogen (((4-cyanophenyl)amino)(4-(dimethylamino)phenyl)methyl)phosphonate (26)
- Phenyl hydrogen (((4-cyanophenyl)amino)(4-(diethylamino)phenyl)methyl) phosphonate (27)
- Phenyl hydrogen ((4-(dimethylamino)phenyl)(phenylamino)methyl)phosphonate (28)
3.3. In Vitro Antifungal Activity
3.4. Evaluation of Cytotoxic Activity
3.4.1. Cell Culture
3.4.2. MTT Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bongomin, F.; Gago, S.; Oladele, R.O.; Denning, D.W. Global and Multi-National Prevalence of Fungal Diseases-Estimate Precision. J. Fungi 2017, 3, 57. [Google Scholar] [CrossRef] [PubMed]
- Konsoula, A.; Tsioutis, C.; Markaki, I.; Papadakis, M.; Agouridis, A.P.; Spernovasilis, N. Lomentospora prolificans: An Emerging Opportunistic Fungal Pathogen. Microorganisms 2022, 10, 1317. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Tudela, J.L.; Berenguer, J.; Guarro, J.; Kantarcioglu, A.S.; Horre, R.; de Hoog, G.S.; Cuenca-Estrella, M. Epidemiology and outcome of Scedosporium prolificans infection, a review of 162 cases. Med. Mycol. 2009, 47, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Konsoula, A.; Agouridis, A.P.; Markaki, L.; Tsioutis, C.; Spernovasilis, N. Lomentospora prolificans Disseminated Infections: A Systematic Review of Reported Cases. Pathogens 2023, 12, 67. [Google Scholar] [CrossRef]
- World Health Organization. WHO Fungal Priority Pathogens List to Guide Research, Development and Public Health Action; WHO: Geneva, Switzerland, 2022; Available online: https://www.who.int/publications/i/item/9789240060241 (accessed on 25 October 2022).
- Houšť, J.; Spížek, J.; Havlíček, V. Antifungal Drugs. Metabolites 2020, 10, 106. [Google Scholar] [CrossRef]
- Ramirez-Garcia, A.; Pellon, A.; Rementeria, A.; Buldain, I.; Barreto-Bergter, E.; Rollin-Pinheiro, R.; Vieira de Meirelles, J.; Xisto, M.I.; Ranque, S.; Havlicek, V.; et al. Scedosporium and Lomentospora: An updated overview of underrated opportunists. Med. Mycol. 2018, 56, S102–S125. [Google Scholar] [CrossRef]
- Mucha, A.; Kafarski, P.; Berlicki, Ł. Remarkable Potential of the α-Aminophosphonate/Phosphinate Structural Motif in Medicinal Chemistry. J. Med. Chem. 2011, 54, 5955–5980. [Google Scholar] [CrossRef]
- Kafarski, P.; Lejczak, B. Biological Activity of Aminophosphonic Acids. Phosphorus Sulfur Silicon Relat. Elem. 1991, 63, 193–215. [Google Scholar] [CrossRef]
- Kafarski, P. Aminophosphonic Acids of Potential Medical Importance. Curr. Med. Chem. Agents 2001, 1, 301–312. [Google Scholar] [CrossRef]
- Keglevich, G.; Bálint, E. The Kabachnik–Fields Reaction: Mechanism and Synthetic Use. Molecules 2012, 17, 12821–12835. [Google Scholar] [CrossRef]
- Kafarski, P.; Górniak, M.; Andrasiak, I. Kabachnik-Fields Reaction Under Green Conditions—A Critical Overview. Curr. Green Chem. 2015, 2, 218–222. [Google Scholar] [CrossRef]
- Manabe, K.; Kobayashi, S. Facile synthesis of α-amino phosphonates in water using a Lewis acid–surfactant-combined catalyst. Chem. Commun. 2000, 8, 669–670. [Google Scholar] [CrossRef]
- Qian, C.; Huang, T.J. One-Pot Synthesis of α-amino Phosphonates from Aldehydes Using Lanthanide Triflate as a Catalyst. J. Org. Chem. 1998, 63, 4125–4128. [Google Scholar] [CrossRef]
- Frouzabadi, H.; Iranpoor, H.; Sobhani, S. Metal Triflate-Catalyzed One-Pot Synthesis of α-Aminophosphonates from Carbonyl Compounds in the Absence of Solvent. Synthesis 2004, 16, 2692–2696. [Google Scholar] [CrossRef]
- Ranu, B.; Hajra, A.; Jana, U. General Procedure for the Synthesis of α-Amino Phosphonates from Aldehydes and Ketones Using Indium(III) Chloride as a Catalyst. Org. Lett. 1999, 1, 1141–1143. [Google Scholar] [CrossRef]
- Lee, S.; Park, J.; Kang, J.; Lee, K. Lanthanide triflate-catalyzed three component synthesis of α-amino phosphonates in ionic liquids. A catalyst reactivity and reusability study. J. Chem. Commun. 2001, 17, 1698–1699. [Google Scholar] [CrossRef]
- Xu, F.; Luo, Y.; Deng, M.; Shen, Q. One-Pot Synthesis of α-Amino Phosphonates Using Samarium Diiodide as a Catalyst Precursor. Eur. J. Org. Chem. 2003, 24, 4728–4730. [Google Scholar] [CrossRef]
- Manjula, A.; Rao, B.; Neelakantan, P. One-Pot Synthesis of α-Aminophosphonates: An Inexpensive Approach. Synth. Commun. 2003, 33, 2963–2969. [Google Scholar] [CrossRef]
- Joly, G.; Jacobsen, E. Thiourea-Catalyzed Enantioselective Hydrophosphonylation of Imines: Practical Access to Enantiomerically Enriched α-Amino Phosphonic Acids. J. Am. Chem. Soc. 2004, 126, 4102–4103. [Google Scholar] [CrossRef]
- Kudrimoti, S.; Bommena, V.R. (Bromodimethyl)sulfonium bromide: An inexpensive reagent for the solvent-free, one-pot synthesis of α-aminophosphonates. Tetrahedron Lett. 2005, 7, 1209–1210. [Google Scholar] [CrossRef]
- Zhan, Z.; Yang, R.; Li, J. Microwave-assisted One-pot Synthesis of α-Amino Phosphonates via Three Component Coupling on a Silica Gel Support. Chem. Lett. 2005, 34, 1042–1043. [Google Scholar] [CrossRef]
- Zefirov, N.S.; Matveeva, E.D. Catalytic Kabachnik-Fields reaction: New horizons for old reaction. Arkivoc 2008, 1, 1–17. [Google Scholar] [CrossRef]
- Xue-Jun, M.; Mao-Yi, L.; Jian-Ping, Z.; Wei, Z. Microwave-assisted solvent-free and catalyst-free Kabachnik–Fields reactions for α-amino phosphonates. Tetrahedron Lett. 2006, 47, 1125–1127. [Google Scholar] [CrossRef]
- Rostamnia, S.; Hassankhani, A. Application of biodegradable supramolecular polymer-supported catalyst for multicomponent synthesis of a-aminophosphonates Kabachnik–Fields reaction. Supramol. Chem. 2014, 26, 736–739. [Google Scholar] [CrossRef]
- Bahadi, R.; Berredjem, M.; Redjemia, R.; Bouacida, S.; Amamra, R.; Meziani, O.; Zerrad, C.; Laichi, Y.; Bachari, K.; Ibrahim-Ouali, M.; et al. A green and novel method for the preparation of a-aminophosphonates using eggshell as catalyst. X-ray study. Phosphorus Sulfur Silicon Relat. Elem. 2022, 197, 1248–1254. [Google Scholar] [CrossRef]
- Shilpa, T.; Harry, N.A.; Ujwaldev, S.M.; Anikumar, G. An Overview of Microwave-Assisted Kabachnik-Fields Reactions. ChenistrySelect 2020, 5, 4422–4436. [Google Scholar] [CrossRef]
- Kabachnik, M.M.; Zobnina, E.V.; Beletskaya, I. Catalyst-Free Microwave-Assisted Synthesis of α-Aminophosphonates in aThree-Component System: R1C(O)R2–(EtO)2P(O)H–RNH2. Synlett 2005, 9, 1393–1396. [Google Scholar] [CrossRef]
- Prauda, I.; Greiner, I.; Ludányi, K.; Keglevich, G. Efficient Synthesis of Phosphono- and Phosphinoxidomethylated N-Heterocycles under Solvent-Free Microwave Conditions. Synth. Commun. 2007, 37, 317–322. [Google Scholar] [CrossRef]
- Keglevich, G.; Szekrénya, A. Eco-Friendly Accomplishment of the Extended Kabachnik–Fields Reaction; a Solvent- and Catalyst-Free Microwave-Assisted Synthesis of α-Aminophosphonates and α-Aminophosphine Oxides. Lett. Org. Chem. 2008, 5, 616–622. [Google Scholar] [CrossRef]
- Bálint, E.; Takács, J.; Drahos, L.; Juranovič, A.; Kočevar, M.; Keglevich, G. α-Aminophosphonates and α-Aminophosphine Oxides by the Microwave-Assisted Kabachnik–Fields Reactions of 3-Amino-6-methyl-2 H-pyran-2-ones. Heteroat. Chem. 2013, 24, 221–225. [Google Scholar] [CrossRef]
- Bálint, E.; Fazekas, E.; Kóti, J.; Keglevich, G. Synthesis of N,N-Bis(dialkoxyphosphinoylmethyl)- and N,N-Bis(diphenylphosphinoylmethyl)-β- and γ-amino acid Derivatives by the Microwave-Assisted Double Kabachnik–Fields Reaction. Heteroat. Chem. 2015, 26, 106–115. [Google Scholar] [CrossRef]
- Bálint, E.; Tajti, Á.; Ladányi-Pára, K.; Tóth, N.; Mátravölgyi, B.; Keglevich, G. Continuous flow synthesis of α-aryl-α-aminophosphonates. Pure Appl. Chem. 2019, 91, 67–76. [Google Scholar] [CrossRef]
- Tajti, Á.; Szatmári, E.; Perdih, F.; Keglevich, G.; Bálint, E. Microwave-Assisted Kabachnik–Fields Reaction with Amino Alcohols as the Amine Component. Molecules 2019, 24, 1640. [Google Scholar] [CrossRef] [PubMed]
- Cordero-Díaz, A.; Robledo-Leal, E.; Hernández-Fernández, E.; Hernández-Núñez, E.; Elizondo-Zertuche, M.; López-Cortina, S.T. Novel α-Aminophosphonates and α-Aminophosphonic Acids: Synthesis, Molecular Docking and Evaluation of Antifungal Activity against Scedosporium Species. Molecules 2022, 27, 3886. [Google Scholar] [CrossRef] [PubMed]
- Pellon, A.; Ramirez-Garcia, A.; Buldain, I.; Antoran, A.; Martin-Souto, L.; Rementeria, A.; Hernando, F.L. Pathobiology of Lomentospora prolificans: Could this species serve as a model of primary antifungal resistance? Int. J. Antimicrob. Agents 2018, 51, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Wallace, T.C.; Giusti, M.M. Evaluation of Parameters the Affect the 4-Dimethylaminocinamaldehyde Assay for Flavanols and Proanthocyanidins. J. Food Sci. 2010, 75, C619–C625. [Google Scholar] [CrossRef]
- Wu, M.; Liu, R.; Wan, D. Convenient One-Pot Synthesis of α-Amino Phosphonates in Water Usingp-Toluenesulfonic Acid as Catalyst for the Kabachnik-Fields Reaction. Heteroat. Chem. 2013, 24, 110–115. [Google Scholar] [CrossRef]
- Ando, K.; Egami, T. Facile synthesis of α-amino phosphonates in water by Kabachnik-Fields reaction using magnesium dodecyl sulfate. Heteroat. Chem. 2011, 22, 358–362. [Google Scholar] [CrossRef]
- Fang, D.; Jiao, C.; Ni, C. SO3H-functionalized ionic liquids catalyzed the synthesis of α-aminophosphonates in aqueous media. Heteroat. Chem. 2010, 21, 546–550. [Google Scholar] [CrossRef]
- Sabo, J.A.; Abdel-Rahman, S.M. Voriconazole: A new triazole antifungal. Ann. Pharmacother. 2000, 34, 1032–1043. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Cob-Calan, N.N.; Chi-Uluac, L.A.; Ortiz-Chi, F.; Cerqueda-García, D.; Navarrete-Vázquez, G.; Ruiz-Sánchez, E.; Hernández-Núñez, E. Molecular docking and dynamics simulation of protein β-tubulin and antifungal cyclic lipopeptides. Molecules 2019, 24, 3387. [Google Scholar] [CrossRef] [PubMed]
- Elizondo-Zertuche, M.; Montoya, A.M.; Robledo-Leal, E.; Garza-Veloz, I.; Sánchez-Nuñez, A.L.; Ballesteron-Elizondo, R.; González, G.M. Comparative Pathogenicity of Lomentospora prolificans (Scedosporium prolificans) Isolates from Mexican Patients. Mycopathologia 2017, 182, 681–689. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comp | JH-P | JC-P | Comp | JH-P | JC-P | Comp | JH-P | JC-P | Comp | JH-P | JC-P |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 5 | 25.85 | 156.26 | 11 | 23.73 | 156.74 | 17 | 24.6 | 142.97 | 23 | 20.36 | 142.24 |
| 6 | 25.72 | 157.1 | 12 | 25.12 | 164.28 | 18 | 22.2 | 140.86 | 24 | 23.6 | 144.0 |
| 7 | 23.5 | 157.07 | 13 | 25.36 | 156.82 | 19 | 21.31 | 138.44 | 25 | 24.26 | 142.4 |
| 8 | 23.31 | 157.47 | 14 | 24.37 | 157.53 | 20 | 20.62 | 140.06 | 26 | 24.25 | 143.34 |
| 9 | 24.02 | 162.6 | 15 | 23.65 | 157.9 | 21 | 21.23 | 157.7 | 27 | 21.98 | 142.1 |
| 10 | 26.11 | 155.71 | 16 | 23.44 | 158.11 | 22 | 23.39 | 142.8 | 28 | 20.36 | 142.24 |
| Strain | MIC100 Range (μg/mL) Compound 7 | MIC100 Range (μg/mL) Compound 11 | MIC100 Range (μg/mL) Compound 13 | MIC100 Range (μg/mL) Compound 22 | MIC100 Range (μg/mL) Compound 27 | MIC100 Range (μg/mL) VRC |
|---|---|---|---|---|---|---|
| L. prolificans (5) | 900->900 | 900->900 | 900->900 | 900->900 | 900->900 | 16->16 |
| Compound (1000 µM) | Viability (%) |
|---|---|
| 7 | 39.01 ± 2.45 |
| 11 | 38.72 ± 1.31 |
| 13 | 41.85 ± 3.13 |
| 22 | 67.91 ± 4.85 |
| 27 | 60.26 ± 3.15 |
| Voriconazole | 68.55 ± 2.05 |
| Etoposide * | 40.79 ± 2.56 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez-Campos, Z.; Elizondo-Zertuche, M.; Hernández-Núñez, E.; Hernández-Fernández, E.; Robledo-Leal, E.; López-Cortina, S.T. Microwave-Assisted Synthesis of Aminophosphonic Derivatives and Their Antifungal Evaluation against Lomentospora prolificans. Molecules 2023, 28, 3995. https://doi.org/10.3390/molecules28103995
Martínez-Campos Z, Elizondo-Zertuche M, Hernández-Núñez E, Hernández-Fernández E, Robledo-Leal E, López-Cortina ST. Microwave-Assisted Synthesis of Aminophosphonic Derivatives and Their Antifungal Evaluation against Lomentospora prolificans. Molecules. 2023; 28(10):3995. https://doi.org/10.3390/molecules28103995
Chicago/Turabian StyleMartínez-Campos, Zuleyma, Mariana Elizondo-Zertuche, Emanuel Hernández-Núñez, Eugenio Hernández-Fernández, Efrén Robledo-Leal, and Susana T. López-Cortina. 2023. "Microwave-Assisted Synthesis of Aminophosphonic Derivatives and Their Antifungal Evaluation against Lomentospora prolificans" Molecules 28, no. 10: 3995. https://doi.org/10.3390/molecules28103995
APA StyleMartínez-Campos, Z., Elizondo-Zertuche, M., Hernández-Núñez, E., Hernández-Fernández, E., Robledo-Leal, E., & López-Cortina, S. T. (2023). Microwave-Assisted Synthesis of Aminophosphonic Derivatives and Their Antifungal Evaluation against Lomentospora prolificans. Molecules, 28(10), 3995. https://doi.org/10.3390/molecules28103995