
The History of Desulfovibrio gigas Aldehyde Oxidoreductase—A Personal View


Abstract
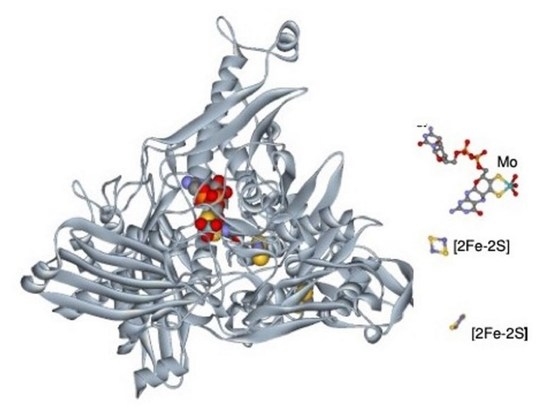
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Context
2. Framing the History
3. The Starting Point and Developments
4. Developing Collaborations and Revealing the Properties of the Metal Sites
5. The 3D Structure We Were Waiting For!
6. After the Structure: Further Insights into Catalysis, Inhibition, and Reactivity
7. Electron-Transfer Complexes
8. Other AORs Isolated from Sulfate Reducers
9. Magnetic Interaction between the Redox Centers of AOR (and XO)
10. Catalysis in Non-Aqueous Solvents
11. Oxygen Atom Insertion vs. Abstraction
12. From Basic to Applied Research: From Bacteria to Humans
13. Epilogue
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moura, J.J.G.; Xavier, A.V.; Bruschi, M.; Le Gall, J.; Hall, D.O.; Cammack, R. A molybdenum-containing iron-sulphur protein from Desulphovibrio gigas. Biochem. Biophys. Res. Commun. 1976, 72, 782–789. [Google Scholar] [CrossRef]
- Moura, J.J.G.; Barata, B.A.S. Aldehyde oxidoreductases and other molybdenum-containing enzymes. Methods Enzymol. 1994, 243, 24–42. [Google Scholar]
- Moura, J.J.G.; Xavier, A.V.; Bruschi, M.; Legall, J.; Cabral, J.M.P. In Proceedings of the 2nd Climax Conferences; Mitchell, P.C.H., Ed.; University of Reading: Reading, UK, 1976. [Google Scholar]
- Turner, N.; Barata, B.; Bray, R.C.; Deistung, J.; Le Gall, J.; Moura, J.J.G. The molybdenum iron-sulphur protein from Desulfovibrio gigas as a form of aldehyde oxidase. Biochem. J. 1987, 243, 755–761. [Google Scholar] [CrossRef]
- Bray, R.C.; Turner, N.A.; Le Gall, J.; Barata, B.A.S.; Moura, J.J.G. Information from epr spectroscopy on the iron-sulphur centres of the iron-molybdenum protein (aldehyde oxidoreductase) of Desulfovibrio gigas. Biochem. J. 1991, 280, 817–820. [Google Scholar] [CrossRef] [PubMed]
- Moura, J.J.G.; Xavier, A.V.; Cammack, R.; Hall, D.O.; Bruschi, M.; Le Gall, J. Oxidation-reduction studies of the Mo-(2Fe-2S) protein from Desulfovibrio gigas. Biochem. J. 1978, 173, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Barata, B.A.S.; Liang, J.; Moura, I.; LeGall, J.; Moura, J.J.G.; Hanh Huynh, B. Mössbauer study of the native, reduced and substrate-reacted Desulfovibrio gigas aldehyde oxido-reductase. European journal of biochemistry. Eur. J. Biochem. 1992, 204, 773–778. [Google Scholar] [CrossRef]
- Hille, R.; Hagen, W.R.; Dunham, W.R. Spectroscopic studies on the iron-sulfur centers of milk xanthine oxidase. J. Biol. Chem. 1985, 260, 10569–10575. [Google Scholar] [CrossRef] [PubMed]
- Zhelyaskov, V.; Yue, K.T.; LeGall, J.; Barata, B.A.S.; Moura, J.J.G. Resonance Raman study on the iron-sulfur centers of Desulfovibrio gigas aldehyde oxidoreductase. Biochim. Biophys. Acta 1995, 1252, 300–304. [Google Scholar] [CrossRef]
- Cramer, S.P.; Moura, J.J.G.; Xavier, A.V.; LeGall, J. Molybdenum EXAFS of the Desulfovibrio gigas Mo(2Fe-2S) protein--structural similarity to "desulfo" xanthine dehydrogenase. Inorg. Biochem. 1984, 20, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Moura, J.J.G. O Ano das Estruturas 3D de Metaloproteínas-1995. Quimica 1996, 63, 35–42. [Google Scholar]
- Tsukihara, T.; Aoyama, H.; Yamashita, E.; Tomizaki, T.; Yamaguchi, H.; Ttoh, K.S.; Nakashima, R.; Yaono, R.; Yoshikawa, S. Structure analysis of bovine heart cytochrome c oxidase at 2.8 A resolution. Science 1995, 269, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Iwata, S.; Ostermeier, C.; Ludwig, B.; Michel, H. Structure at 2.8 Å resolution of cytochrome c oxidase from Paracoccus denitrificans. Title. Nature 1995, 376, 660–669. [Google Scholar] [CrossRef]
- Fulop, V.; Ridout, C.J.; Greenwood, C.; Hajdu, J. Crystal structure of the di-haem cytochrome c peroxidase from Pseudomonas aeruginosa. Structure 1995, 3, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Fulop, V.; Moir, J.B.W.; Ferguson, S.J.; Hajdu, J. Crystallization and preliminary crystallographic study of cytochrome-cd1 nitrite reductase from Thiosphaera pantotropha. J. Mol. Biol. 1993, 232, 1211–1212. [Google Scholar] [CrossRef] [PubMed]
- Jabri, E.; Carr, M.B.; Hausinger, R.P.; Karplus, P.A. The crystal structure of urease from Klebsiella aerogenes. Science 1995, 268, 998–1004. [Google Scholar] [CrossRef]
- Crane, B.R.; Siegel, L.M.; Getzoff, E.D. Sulfite reductase structure at 1.6 A: Evolution and catalysis for reduction of inorganic anions. Science 1995, 270, 59–67. [Google Scholar]
- Volbeda, A.; Charon, M.H.; Piras, C.; Hatchikian, C.E.C.; Frey, M.; Fontecilla-Camps, J.C. Crystal structure of the nickel-iron hydrogenase from Desulfovibrio gigas. Nature 1995, 373, 580–587. [Google Scholar] [CrossRef]
- Logan, D.T.; Su, X.D.; Aberg, K.; Regnstriim, A.K.; Hajdu, J.; Eklund, H.; Nordlund, P. Crystal Structure of Reduced Protein R2 of Ribonucleotide Reductase. Structure 1995, 4, 1053–1064. [Google Scholar] [CrossRef]
- Chan, M.K.; Mukund, S.; Kletzin, A.; Adam, M.W.W.; Rees, C.D. Structure of a Hyperthermophilic Tungstopterin Enzyme, Aldehyde Ferredoxin Oxido-reductase. Science 1995, 267, 1463–1469. [Google Scholar] [CrossRef]
- Staler, N.; Klabunde, T.; Tucker, P.; Witzel, H.; Krebs, B. Crystal structure of a purple acid phosphatase containing a dinuclear Fe(III)-Zn(II) active site. Science 1995, 268, 1489–1492. [Google Scholar]
- Schindelin, H.; Kisker, C.; Hilton, I.; Rojagopalan, K.; Rees, C.D. Crystal Structure of DMSO Reductase: Redox-Linked Chan- ges in Molybdopterin Coordination. Science 1996, 272, 1615–1621. [Google Scholar] [CrossRef] [PubMed]
- Gladyshev, V.N.; Boyington, I.C.; Khangulov, S.V.; Gtaham, D.A.; Stadtman, T.C.; Sun, P.D. Crystal structure of formate dehydrogenase H: Catalysis involving Mo, molybdopterin, selenocysteine, and an Fe4S4 cluster. J. Biol. Chem. 1996, 27, 8095–8100. [Google Scholar] [CrossRef]
- Gennis, G.; Ferguson-Miller, S. Structure of cytochrome c oxidase, energy generator of aerobic life. Science 1995, 269, 1063–1064. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.J.P. Purpose of proton pathways. Nature 1995, 376, 643. [Google Scholar] [CrossRef]
- Cammack, R. Splitting molecular hydrogen. Nature 1995, 373, 556–557. [Google Scholar] [CrossRef] [PubMed]
- Lippard, S.J. At last the crystal structure of urease. Science 1995, 268, 996–997. [Google Scholar] [CrossRef]
- Thoenes, U.; Flores, O.L.; Neves, A.; Devreese, B.; Van Beeumen, J.J.; Huber, R.; Romão, M.J.; LeGall, J.; Moura, J.J.G.; Rodrigues Pousada, C. Molecular cloning, and sequence analysis of the gene of the molybdenum-containing aldehyde oxido-reductase of Desulfovibrio gigas. The deduced amino acid sequence shows similarity to xanthine dehydrogenase. Eur. J. Biochem. 1994, 220, 901–910. [Google Scholar] [CrossRef]
- Morais-Silva, F.O.; Rezende, A.M.; Pimentel, C.; Santos, C.I.; Clemente, C.; Varela–Raposo, A.; Resende, D.M.; Silva, S.M.; Oliveira, L.M.; Matos, M.; et al. Genome sequence of the model sulfate reducer Desulfovibrio gigas: A comparative analysis within the Desulfovibrio genus. MicrobiologyOpen 2014, 3, 513–530. [Google Scholar] [CrossRef]
- Romão, M.J.; Barata, B.A.S.; Archer, M.; Lobeck, K.; Moura, I.; Carrondo, M.A.; LeGall, J.; Lottspeich, F.; Huber, R.; Moura, J.J.G. Subunit composition, crystallization and preliminary crystallographic studies of the Desulfovibrio gigas aldehyde oxidoreductase containing molybdenum and [2Fe-2S] centers. Eur. J. Biochem. 1993, 215, 729–732. [Google Scholar] [CrossRef]
- Romão, M.J.; Archer, M.; Moura, I.; Moura, J.J.G.; LeGall, J.; Engh, R.; Schneider, M.; Hof, P.; Huber, R. Crystal structure of the xanthine oxidase-related aldehyde oxido-reductase from D. gigas. Science 1995, 270, 1170–1176. [Google Scholar] [CrossRef]
- Rebelo, J.; Dias, J.; Huber, R.; Moura, J.; Romão, M.J. J. Structure refinement of the aldehyde oxidoreductase from Desulfovibrio gigas (MOP) at 1.28 Å. J. Biol. Inorg. Chem. 2001, 6, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Enroth, C.; Eger, B.T.; Okamoto, K.; Nishino, T.; Nishino, T.; Pai, E.F. Purification, crystallization and preliminary X-ray diffraction studies of xanthine dehydrogenase and xanthine oxidase isolated from bovine milk. Proc. Natl. Acad. Sci. USA 2000, 97, 10723. [Google Scholar] [CrossRef]
- Huber, R.; Hof, P.; Duarte, R.O.; Moura, J.J.G.; Moura, I.; Liu, M.-; LeGall, J.; Hille, R.; Archer, M.; Romão, M.J. A structure-based catalytic mechanism for the xanthine oxidase family of molybdenum enzymes. Proc. Natl. Acad. Sci. USA 1996, 93, 8846–8851. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Kusano, T.; Nishino, T. Chemical Nature and Reaction Mechanisms of the Molybdenum Cofactor of Xanthine Oxidoreductase. Curr. Pharm. Des. 2013, 19, 2606–2614. [Google Scholar] [CrossRef]
- Hille, R.; Nishino, T.; Bittner, F. Molybdenum enzymes in higher organisms. Coord. Chem. Rev. 2011, 255, 1179–1205. [Google Scholar] [CrossRef]
- Hille, R.; Hall, J.; Basu, P. The Mononuclear Molybdenum Enzymes. Chem. Rev. 2014, 114, 3963–4038. [Google Scholar] [CrossRef]
- Fernandes, H.; Maia, L.B.; Ribeiro, P.M.; Moura, J.J.G.; Cerqueira, N.M. The complete catalytic mechanism of Xanthine Oxidase: A computational study. Inorg. Chem. Front. 2021, 8, 405. [Google Scholar]
- Santos-Silva, T.; Ferroni, F.; Thapper, A.; Marangon, J.; González, P.J.; Rizzi, A.C.; Moura, I.; Moura, J.J.G.; Romão, M.J.; Brondino, C.D. Kinetic, structural, and EPR studies reveal that aldehyde oxidoreductase from Desulfovibrio gigas does not need a sulfido ligand for catalysis and give evidence for a direct Mo-C interaction in a biological system. J. Am. Chem. Soc. 2009, 131, 7990–7998. [Google Scholar] [CrossRef]
- Correia, H.D.; Marangon, J.; Brondino, C.D.; Moura, J.J.G.; Romão, M.J.; González, P.J.; Santos-Silva, T. Aromatic aldehydes at the active site of aldehyde oxidoreductase from Desulfovibrio gigas: Reactivity and molecular details of the enzyme-substrate and enzyme-product interaction. J. Biol. Inorg. Chem. 2015, 20, 219–229. [Google Scholar] [CrossRef]
- Marangon, J.; Correia, H.D.; Brondino, C.D.; Moura, J.J.G.; Romão, M.J.; González, P.J.; Santos-Silva, T. Kinetic and structural studies of aldehyde oxidoreductase from Desulfovibrio gigas reveal a dithiolene-based chemistry for enzyme activation and inhibition by H(2)O(2). PLoS ONE 2013, 8, e83234. [Google Scholar] [CrossRef]
- Boer, D.R.; Thapper, A.; Brondino, C.D.; Romão, M.J.; Moura, J.J.G. X-ray crystal structure and EPR spectra of “arsenite-inhibited” Desulfovibriogigas aldehyde dehydrogenase: A member of the xanthine oxidase family. J. Am. Chem. Soc. 2004, 126, 8614–8615. [Google Scholar] [CrossRef]
- Thapper, A.; Boer, D.R.; Brondino, C.D.; Moura, J.J.G.; Romão, M.J. Correlating EPR and X-ray structural analysis of arsenite-inhibited forms of aldehyde oxidoreductase. J. Biol. Inorg. Chem. 2007, 12, 353–366. [Google Scholar] [CrossRef]
- Santos, M.M.C.; Sousa, P.M.P.; Goncalves, M.L.S.; Romao, M.J.R.; Moura, I.; Moura, J.J.G. Direct electrochemistry of the Desulfovibrio gigas aldehyde oxidoreductase. Eur. J. Biochem. 2004, 271, 1329–1338. [Google Scholar] [CrossRef]
- Correia, C.; Monzan, E.; Moura, I.; Lampreia, J.; Moura, J.J.G. Cross-linking between cytochrome c3 and flavodoxin from Desulfovibrio gigas. Biochem. Biophys. Res. Commun. 1999, 256, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Barata, B.A.S.; LeGall, J.; Moura, J.J.G. Aldehyde oxidoreductase activity in Desulfovibrio gigas: In vitro reconstitution of an electron-transfer chain from aldehydes to the production of molecular hydrogen. Biochemistry 1993, 32, 11559–11568. [Google Scholar] [CrossRef] [PubMed]
- Palma, P.N.; Krippahl, L.; Wampler, J.E.; Moura, J.J.G. BiGGER: A new (soft) docking algorithm for predicting protein interactions. Proteins 2000, 39, 372–384. [Google Scholar] [CrossRef]
- Krippahl, L.; Moura, J.J.; Palma, P.N. Modeling protein complexes with BiGGER. Proteins 2003, 52, 19–23. [Google Scholar] [CrossRef]
- Krippahl, L.; Palma, P.N.; Moura, I.; Moura, J.J.G. Modelling the electron-transfer complex between aldehyde oxidoreductase and flavodoxin. Eur. J. Inorg. Chem. 2006, 3835–3840. [Google Scholar] [CrossRef]
- Duarte, R.O.; Archer, M.; Dias, J.M.; Bursakov, S.; Huber, R.; Moura, I.; Romão, M.J.; Moura, J.J.G. Biochemical/spectroscopic characterization and preliminary X-ray analysis of a new aldehyde oxidoreductase isolated from Desulfovibrio desulfuricans ATCC 2777. Biochem. Biophys. Res. Commun. 2000, 268, 745–749. [Google Scholar] [CrossRef]
- Rebelo, J.; Macieira, S.; Dias, J.M.; Huber, R.; Ascenso, C.S.; Rusnak, F.; Moura, J.J.G.; Moura, I.; Romão, M.J. Gene sequence and crystal structure of the aldehyde oxidoreductase from Desulfovibrio desulfuricans ATCC 27774. J. Mol. Biol. 2000, 297, 135–146. [Google Scholar] [CrossRef]
- Andrade, S.L.A.; Brondino, C.D.; Feio, M.J.; Moura, I.; Moura, J.J.G. Aldehyde oxidoreductase activity in Desulfovibrio alaskensis NCIMB 13491 EPR assignment of the proximal [2Fe-2S] cluster to the Mo site. Eur. J. Biochem. 2000, 267, 2054–2061. [Google Scholar] [CrossRef]
- Thapper, A.; Rivas, M.G.; Brondino, C.D.; Ollivier, B.; Fauque, G.; Moura, I.; Moura, J.J.G. Biochemical and spectroscopic characterization of an aldehyde oxidoreductase isolated from Desulfovibrio aminophilus. J. Inorg. Biochem. 2006, 100, 44–50. [Google Scholar] [CrossRef]
- Goámez, M.C.; Neuman, N.I.; Dalosto, S.D.; González, P.J.; Moura, J.J.G.; Rizzi, A.C.; Brondino, C.D. Isotropic exchange interaction between Mo and the proximal FeS center in the xanthine oxidase family member aldehyde oxidoreductase from Desulfovibrio gigas on native and polyalcohol inhibited samples: An EPR and QM/MM study. J. Biol. Inorg. Chem. 2015, 20, 233–242. [Google Scholar] [CrossRef] [PubMed]
- González, P.J.; Barrera, G.I.; Rizzi, A.C.; Moura, J.J.G.; Passeggi, M.C.G.; Brondino, C. D EPR studies of the Mo-enzyme aldehyde oxidoreductase from Desulfovibrio gigas: An application of the Bloch-Wangsness-Redfield theory to a system containing weakly-coupled paramagnetic redox centers with different relaxation rate. J. Inorg. Biochem. 2009, 103, 1342–1346. [Google Scholar] [CrossRef] [PubMed]
- Caldeira, J.; Belle, V.; Asso, M.; Guigliarelli, B.; Moura, I.; Moura, J.J.G.; Bertrand, P. Analysis of the electron paramagnetic resonance properties of the [2Fe-2S]1+ centers in molybdenum enzymes of the xanthine oxidase family: Assignment of signals I and II. Biochemistry 2000, 39, 2700–2707. [Google Scholar] [CrossRef]
- More, C.; Asso, M.; Roger, G.; Guigliarelli, B.; Caldeira, J.; Moura, J.J.G.; Bertrand, P. Study of the spin-spin interactions between the metal centers of Desulfovibrio gigas aldehyde oxidoreductase: Identification of the reducible sites of the [2Fe-2S]1+,2+ cluster. Biochemistry 2005, 44, 11628–11635. [Google Scholar] [CrossRef]
- Andrade, S.L.A.; Brondino, C.D.; Kamenskaya, E.O.; Levashov, A.V.; Moura, J.J.G. Kinetic behavior of Desulfovibrio gigas aldehyde oxidoreductase encapsulated in reverse micelles. Biochem. Biophys. Res. Commun. 2003, 308, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Maia, L.B.; Moura, J.J.G. How biology handles nitrite. Chem. Rev. 2014, 114, 5273–5357. [Google Scholar]
- Maia, L.B.; Pereira, V.; Mira, L.; Moura, J.J. G Nitrite reductase activity of rat and human xanthine oxidase, xanthine dehydrogenase, and aldehyde oxidase: Evaluation of their contribution to NO formation in vivo. Biochemistry 2015, 54, 685–710. [Google Scholar] [CrossRef] [PubMed]
- Maia, L.B.; Moura, J.J.G. Putting xanthine oxidoreductase and aldehyde oxidase on the NO metabolism map: Nitrite reduction by molybdoenzyme. Redox Biology 2018, 19, 274–289. [Google Scholar] [CrossRef]
- Maia, L.B.; Moura, J.J.G. Nitrite reduction by molybdoenzymes: A new class of nitric oxide-forming nitrite reductases. J. Biol. Inorg. Chem. 2015, 20, 403–433. [Google Scholar] [CrossRef]
- Wang, J.; Krizowski, S.; Fischer-Schrader, K.; Niks, D.; Tejero, J.; Sparacino-Watkins, C.; Wang, L.; Ragireddy Frizzell, S.; Eric EKelley, E.E.; Zhang, Y.; et al. Sulfite Oxidase Catalyzes Single-Electron Transfer at Molybdenum Domain to Reduce Nitrite to Nitric Oxide. Antioxid. Redox Signal. 2015, 1, 283–294. [Google Scholar] [CrossRef]
- Rockel, P.; Strube, F.; Rockel, A.; Wildt, J.; Kaiser, W.M. Regulation of nitric oxide (NO) production by plant nitrate reductase in vivo and in vitro. J. Exp. Bot. 2002, 53, 103–110. [Google Scholar] [CrossRef]
- Vine, C.E.; Purewal, S.K.; Cole, J.A. NsrR-dependent method for detecting nitric oxide accumulation in the Escherichia coli cytoplasm and enzymes involved in NO production. FEMS Microbiol. Lett. 2011, 325, 108–114. [Google Scholar] [CrossRef]
- Rowley, G.; Hensen, D.; Felgate, H.; Arkenberg, A.; Appia-Ayme, C.; PriorK; Harrington, C.; Field, S.J.; Butt, J.N.; Baggs, E.; et al. Resolving the contributions of the membrane-bound and periplasmic nitrate reductase systems to nitric oxide and nitrous oxide production in Salmonella enterica serovar Typhimurium. Biochem. J. 2012, 441, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.F.; Grainge, M.J.; Zhang, W.; Doherty, M. Impact of gout on the risk of atrial fibrillation. Nat. Rev. Rheumatol. 2015, 11, 649–662. [Google Scholar] [CrossRef]
- Terao, M.; Garattini, E.; Romão, M.J.; Leimkühler, S. Evolution, expression, and substrate specificities of aldehyde oxidase enzymes in eukaryotes. J. Biol. Chem. 2020, 295, 5377–5389. [Google Scholar] [CrossRef] [PubMed]
- Romão, M.J.; Coelho, C.; Santos-Silva, T.; Foti, A.; Terao, M.; Garattini, E.; Leimkühler, S. Structural basis for the role of mammalian aldehyde oxidases in the metabolism of drugs and xenobiotics. Curr. Opin. Chem. Biol. 2017, 37, 39–47. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moura, J.J.G. The History of Desulfovibrio gigas Aldehyde Oxidoreductase—A Personal View. Molecules 2023, 28, 4229. https://doi.org/10.3390/molecules28104229
Moura JJG. The History of Desulfovibrio gigas Aldehyde Oxidoreductase—A Personal View. Molecules. 2023; 28(10):4229. https://doi.org/10.3390/molecules28104229
Chicago/Turabian StyleMoura, José J. G. 2023. "The History of Desulfovibrio gigas Aldehyde Oxidoreductase—A Personal View" Molecules 28, no. 10: 4229. https://doi.org/10.3390/molecules28104229
APA StyleMoura, J. J. G. (2023). The History of Desulfovibrio gigas Aldehyde Oxidoreductase—A Personal View. Molecules, 28(10), 4229. https://doi.org/10.3390/molecules28104229