
Organocatalytic Asymmetric Peroxidation of γ,δ-Unsaturated β-Keto Esters—A Novel Route to Chiral Cycloperoxides

Abstract
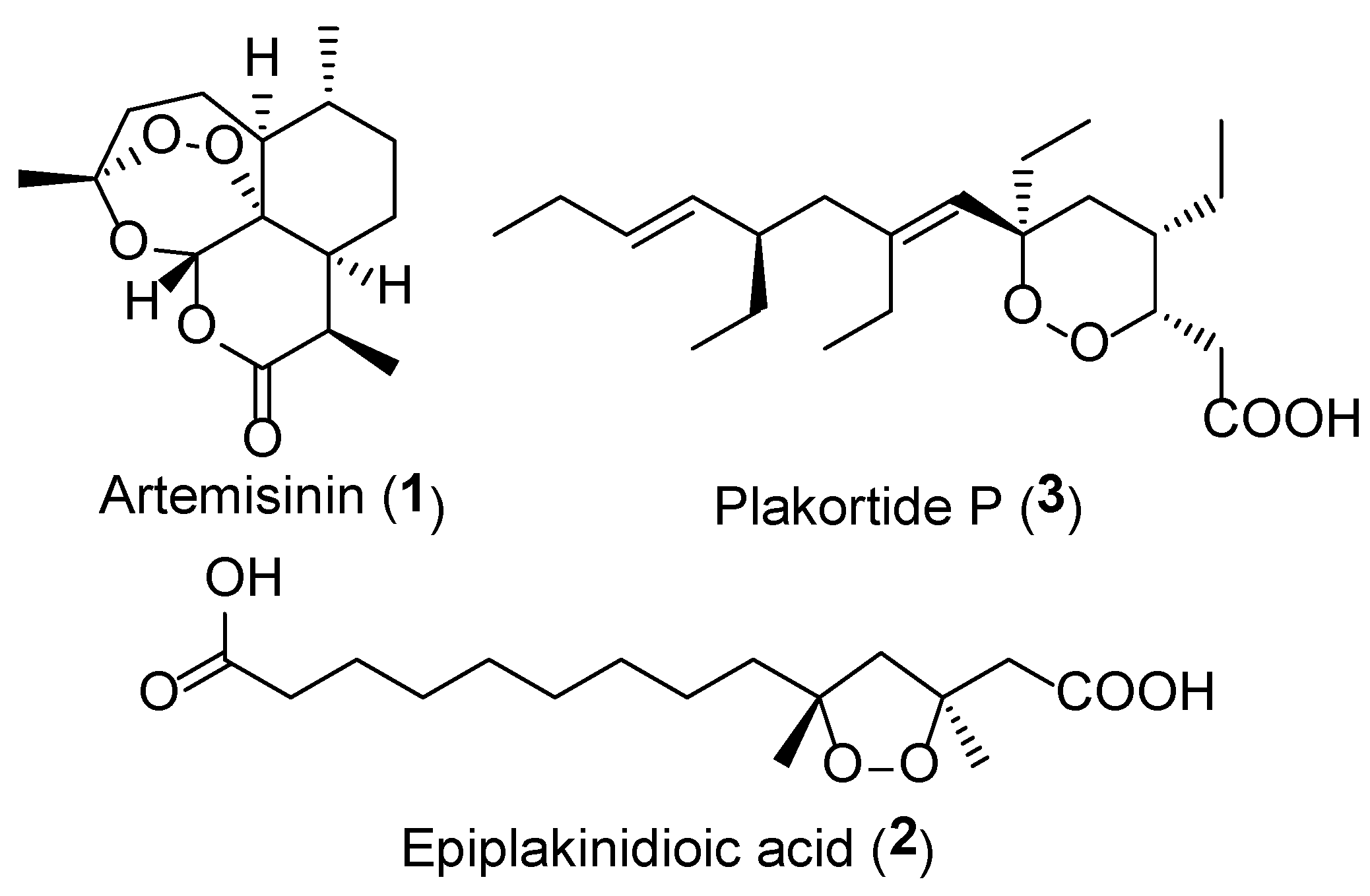
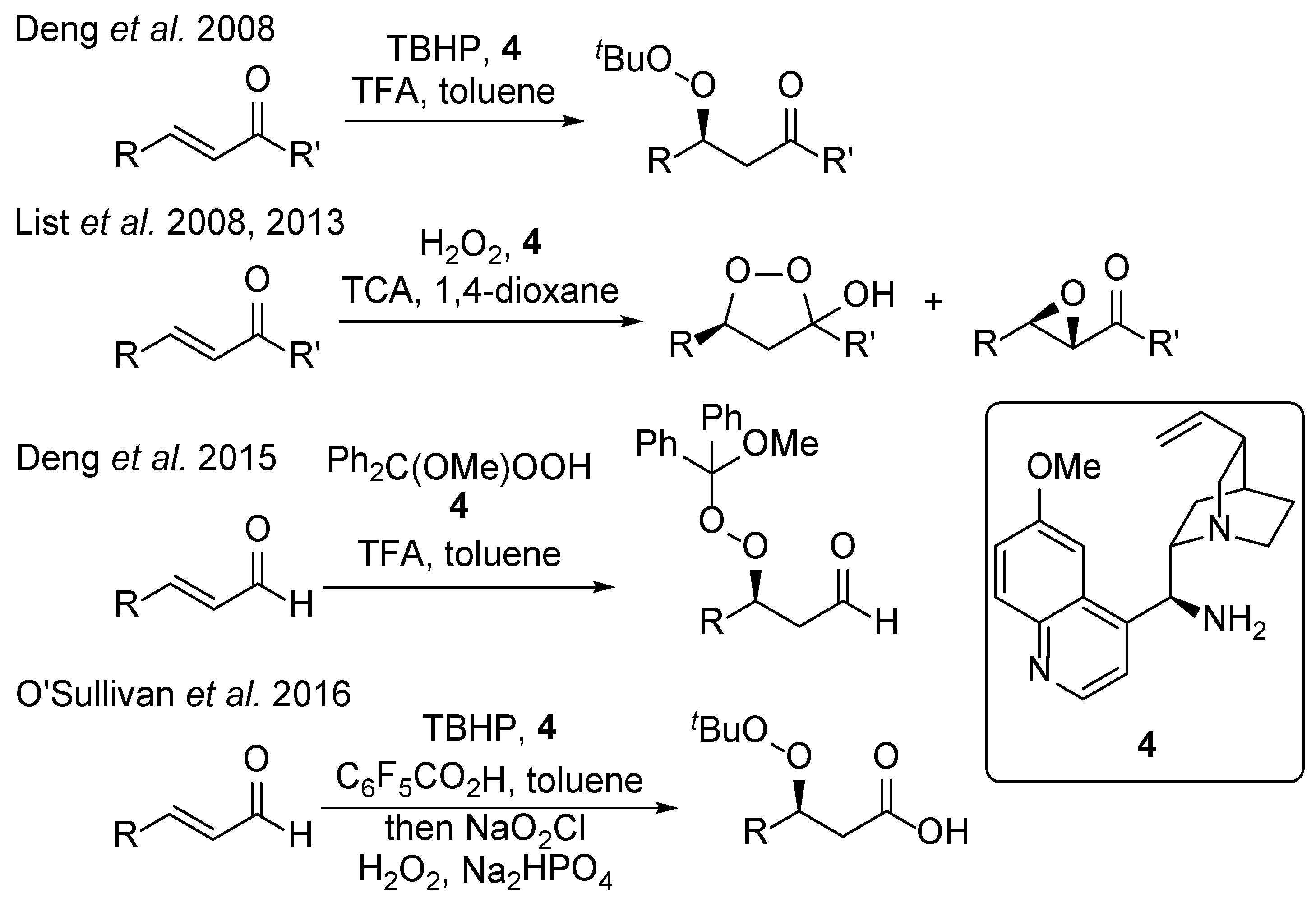
:1. Introduction
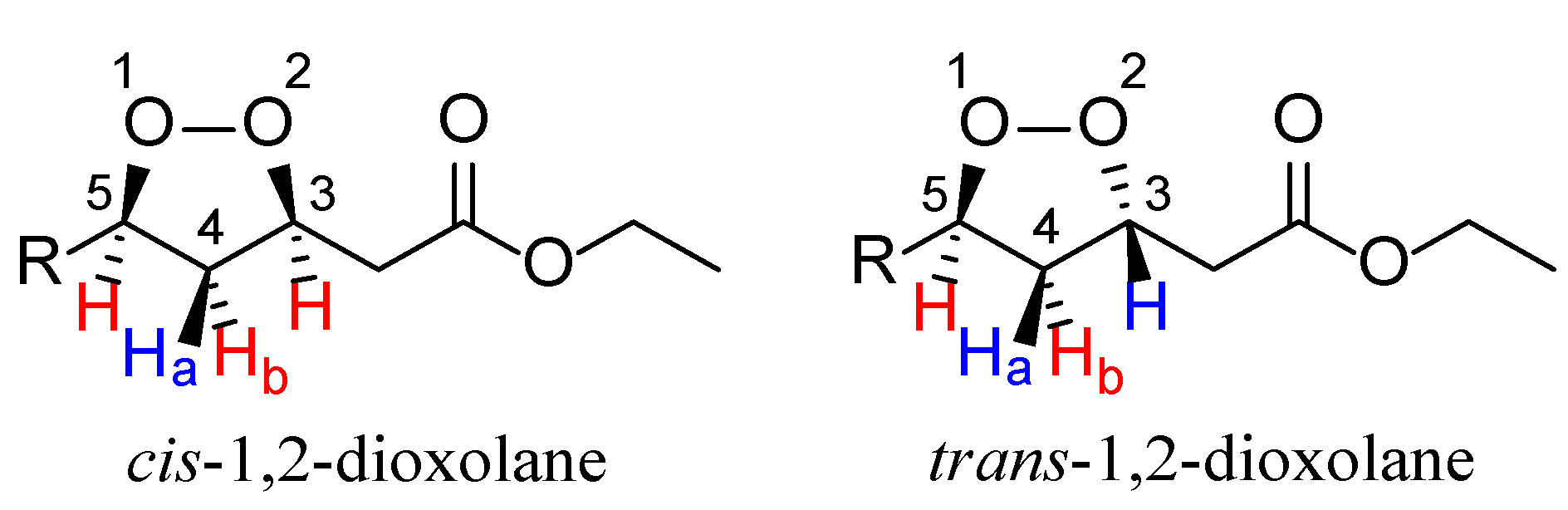
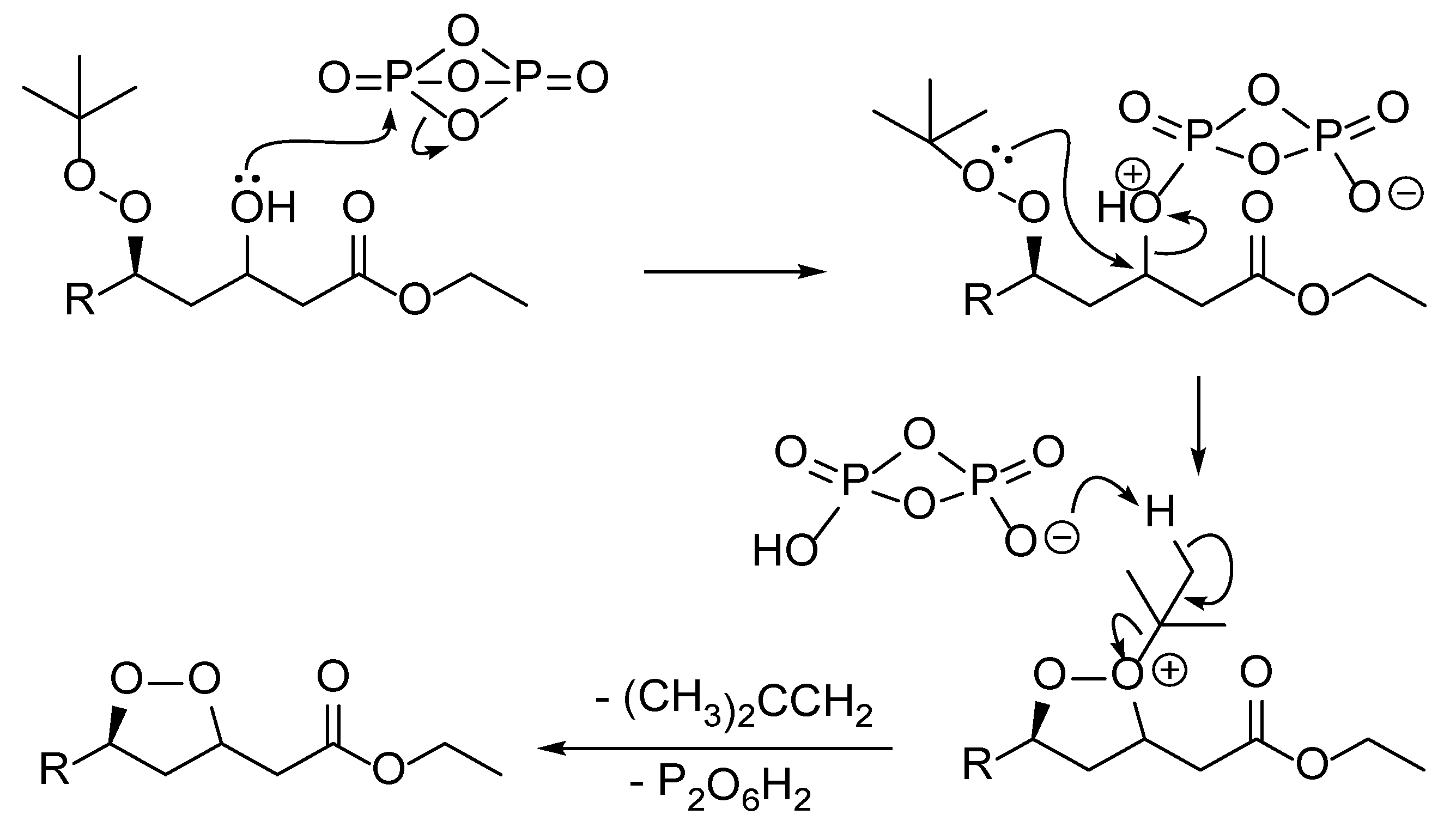
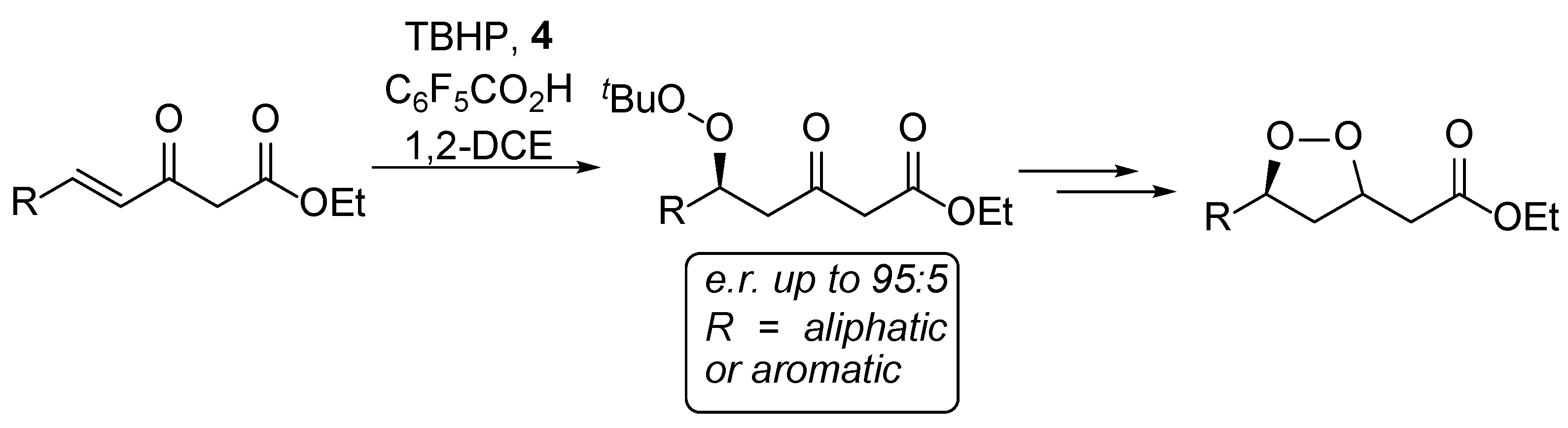
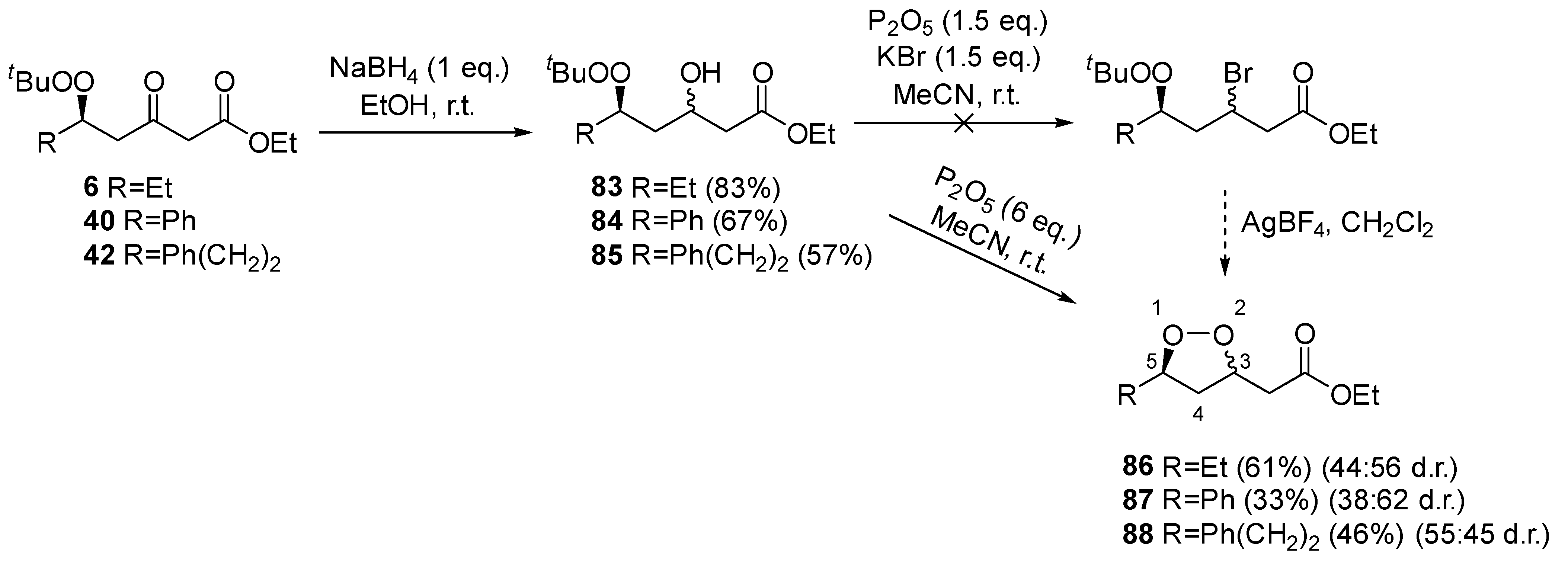
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Casteel, D.A. Peroxy natural products. Nat. Prod. Rep. 1992, 9, 289–312. [Google Scholar] [CrossRef] [PubMed]
- Casteel, D.A. Peroxy natural products. Nat. Prod. Rep. 1999, 16, 55–73. [Google Scholar] [CrossRef]
- Liu, D.-Z.; Liu, J.-K. Peroxy natural products. Nat. Prod. Bioprospecting 2013, 3, 161–206. [Google Scholar] [CrossRef]
- Norris, M.D.; Perkins, M.V. Structural diversity and chemical synthesis of peroxide and peroxide-derived polyketide metabolites from marine sponges. Nat. Prod. Rep. 2016, 33, 861–880. [Google Scholar] [CrossRef]
- Kossuga, M.H.; Nascimento, A.M.; Reimão, J.Q.; Tempone, A.G.; Taniwaki, N.N.; Veloso, K.; Ferreira, A.G.; Cavalcanti, B.C.; Pessoa, C.; Moraes, M.O.; et al. Antiparasitic, Antineuroinflammatory, and Cytotoxic Polyketides from the Marine Sponge Plakortis angulospiculatus Collected in Brazil. J. Nat. Prod. 2008, 71, 334–339. [Google Scholar] [CrossRef]
- Dembitsky, V.M. Bioactive peroxides as potential therapeutic agents. Eur. J. Med. Chem. 2008, 43, 223–251. [Google Scholar] [CrossRef]
- Dembitsky, V.M. Astonishing Diversity of Natural Peroxides as Potential Therapeutic Agents. J. Mol. Genet. Med. 2015, 9, 1000163. [Google Scholar]
- Su, X.-Z.; Miller, L.H. The discovery of artemisinin and the Nobel Prize in Physiology or Medicine. Sci. China Life Sci. 2015, 58, 1175–1179. [Google Scholar] [CrossRef]
- WHO. WHO Guidelines for Malaria; World Health Organisation: Geneva, Switzerland, 2022; Available online: https://www.who.int/publications/i/item/guidelines-for-malaria (accessed on 10 April 2023).
- Chen, Y.; Killday, K.B.; McCarthy, P.J.; Schimoler, R.; Chilson, K.; Selitrennikoff, C.; Pomponi, S.A.; Wright, A.E. Three New Peroxides from the Sponge Plakinastrella Species. J. Nat. Prod. 2001, 64, 262–264. [Google Scholar] [CrossRef]
- Jiménez-Romero, C.; Ortiz, I.; Vicente, J.; Vera, B.; Rodríguez, A.D.; Nam, S.; Jove, R. Bioactive Cycloperoxides Isolated from the Puerto Rican Sponge Plakortis halichondrioides. J. Nat. Prod. 2010, 73, 1694–1700. [Google Scholar] [CrossRef]
- Terent’ev, A.O.; Borisov, D.A.; Vil’, V.A.; Dembitsky, V.M. Synthesis of five- and six-membered cyclic organic peroxides: Key transformations into peroxide ring-retaining products. Beilstein J. Org. Chem. 2014, 10, 34–114. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, H.; O’Reilly, K.; Gupta, M.K.; Horgan, C.; O’Leary, E.M.; O’Sullivan, T.P. Advances in the synthesis of acyclic peroxides. RSC Adv. 2017, 7, 19506–19556. [Google Scholar] [CrossRef]
- Lassaletta, J.M. Spotting trends in organocatalysis for the next decade. Nat. Commun. 2020, 11, 3787. [Google Scholar] [CrossRef] [PubMed]
- Alemán, J.; Cabrera, S. Applications of asymmetric organocatalysis in medicinal chemistry. Chem. Soc. Rev. 2013, 42, 774–793. [Google Scholar] [CrossRef]
- Hughes, D.L. Asymmetric Organocatalysis in Drug Development—Highlights of Recent Patent Literature. Org. Process Res. Dev. 2018, 22, 574–584. [Google Scholar] [CrossRef]
- Grondal, C.; Jeanty, M.; Enders, D. Organocatalytic cascade reactions as a new tool in total synthesis. Nat. Chem. 2010, 2, 167–178. [Google Scholar] [CrossRef]
- Marqués-López, E.; Herrera, R.P.; Christmann, M. Asymmetric organocatalysis in total synthesis—A trial by fire. Nat. Prod. Rep. 2010, 27, 1138–1167. [Google Scholar] [CrossRef]
- Abbasov, M.E.; Romo, D. The ever-expanding role of asymmetric covalent organocatalysis in scalable, natural product synthesis. Nat. Prod. Rep. 2014, 31, 1318–1327. [Google Scholar] [CrossRef]
- Xiang, S.-H.; Tan, B. Advances in asymmetric organocatalysis over the last 10 years. Nat. Commun. 2020, 11, 3786. [Google Scholar] [CrossRef]
- García Mancheño, O.; Waser, M. Recent Developments and Trends in Asymmetric Organocatalysis. Eur. J. Org. Chem. 2023, 26, e202200950. [Google Scholar] [CrossRef]
- Han, B.; He, X.-H.; Liu, Y.-Q.; He, G.; Peng, C.; Li, J.-L. Asymmetric organocatalysis: An enabling technology for medicinal chemistry. Chem. Soc. Rev. 2021, 50, 1522–1586. [Google Scholar] [CrossRef] [PubMed]
- List, B. Introduction: Organocatalysis. Chem. Rev. 2007, 107, 5413–5415. [Google Scholar] [CrossRef]
- MacMillan, D.W.C. The advent and development of organocatalysis. Nature 2008, 455, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Marcelli, T. Organocatalysis: Cinchona catalysts. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 142–152. [Google Scholar] [CrossRef]
- Jiang, L.; Chen, Y.-C. Recent advances in asymmetric catalysis with cinchona alkaloid-based primary amines. Catal. Sci. Technol. 2011, 1, 354–365. [Google Scholar] [CrossRef]
- Marcelli, T.; Hiemstra, H. Cinchona Alkaloids in Asymmetric Organocatalysis. Synthesis 2010, 2010, 1229–1279. [Google Scholar] [CrossRef]
- Lu, X.; Liu, Y.; Sun, B.; Cindric, B.; Deng, L. Catalytic Enantioselective Peroxidation of α,β-Unsaturated Ketones. J. Am. Chem. Soc. 2008, 130, 8134–8135. [Google Scholar] [CrossRef]
- Reisinger, C.M.; Wang, X.; List, B. Catalytic Asymmetric Hydroperoxidation of α,β-Unsaturated Ketones: An Approach to Enantiopure Peroxyhemiketals, Epoxides, and Aldols. Angew. Chem. Int. Ed. 2008, 47, 8112–8115. [Google Scholar] [CrossRef]
- Lifchits, O.; Mahlau, M.; Reisinger, C.M.; Lee, A.; Farès, C.; Polyak, I.; Gopakumar, G.; Thiel, W.; List, B. The Cinchona Primary Amine-Catalyzed Asymmetric Epoxidation and Hydroperoxidation of α,β-Unsaturated Carbonyl Compounds with Hydrogen Peroxide. J. Am. Chem. Soc. 2013, 135, 6677–6693. [Google Scholar] [CrossRef]
- Hu, L.; Lu, X.; Deng, L. Catalytic Enantioselective Peroxidation of α,β-Unsaturated Aldehydes for the Asymmetric Synthesis of Biologically Important Chiral Endoperoxides. J. Am. Chem. Soc. 2015, 137, 8400–8403. [Google Scholar] [CrossRef]
- O’Reilly, K.; Gupta, M.K.; Gandhi, H.K.; Kumar, V.P.; Eccles, K.S.; Lawrence, S.E.; O’Sullivan, T.P. Cinchona-catalysed, Enantioselective Synthesis of β-Peroxycarboxylic Acids, β-Peroxyesters and β-Peroxyalcohols. Curr. Org. Chem. 2016, 20, 2633–2638. [Google Scholar] [CrossRef]
- O’Reilly, K.; Gupta, M.K.; Gandhi, H.; Kumar, V.P.; O’Sullivan, T.P. Asymmetric Peroxidation of α,β-Unsaturated Aldehydes under Diarylprolinol Ether Catalysis. Curr. Org. Chem. 2017, 21, 2013–2016. [Google Scholar] [CrossRef]
- Russo, A.; Lattanzi, A. Catalytic Asymmetric β-Peroxidation of Nitroalkenes. Adv. Synth. Catal. 2008, 350, 1991–1995. [Google Scholar] [CrossRef]
- Lu, X.; Deng, L. Catalytic Asymmetric Peroxidation of α,β-Unsaturated Nitroalkenes by a Bifunctional Organic Catalyst. Org. Lett. 2014, 16, 2358–2361. [Google Scholar] [CrossRef]
- Benetti, S.; Romagnoli, R.; De Risi, C.; Spalluto, G.; Zanirato, V. Mastering beta-Keto Esters. Chem. Rev. 1995, 95, 1065–1114. [Google Scholar] [CrossRef]
- Everaere, K.; Mortreux, A.; Carpentier, J.-F. Ruthenium(II)-Catalyzed Asymmetric Transfer Hydrogenation of Carbonyl Compounds with 2-Propanol and Ephedrine-Type Ligands. Adv. Synth. Catal. 2003, 345, 67–77. [Google Scholar] [CrossRef]
- Ratovelomanana-Vidal, V.; Girard, C.; Touati, R.; Tranchier, J.P.; Hassine, B.B.; Genêt, J.P. Enantioselective Hydrogenation of β-Keto Esters using Chiral Diphosphine-Ruthenium Complexes: Optimization for Academic and Industrial Purposes and Synthetic Applications. Adv. Synth. Catal. 2003, 345, 261–274. [Google Scholar] [CrossRef]
- Bariotaki, A.; Kalaitzakis, D.; Smonou, I. Enzymatic Reductions for the Regio- and Stereoselective Synthesis of Hydroxy-keto Esters and Dihydroxy Esters. Org. Lett. 2012, 14, 1792–1795. [Google Scholar] [CrossRef]
- Heravi, M.M.; Khaghaninejad, S.; Mostofi, M. Chapter one—Pechmann reaction in the synthesis of coumarin derivatives. In Advances in Heterocyclic Chemistry; Academic Press: Cambridge, MA, USA, 2014; Volume 112, pp. 1–50. [Google Scholar] [CrossRef]
- Hennessy, M.C.; O’Sullivan, T.P. Recent advances in the transesterification of β-keto esters. RSC Adv. 2021, 11, 22859–22920. [Google Scholar] [CrossRef]
- Khademi, Z.; Heravi, M.M. Applications of Claisen condensations in total synthesis of natural products. An old reaction, a new perspective. Tetrahedron 2022, 103, 132573. [Google Scholar] [CrossRef]
- Govender, T.; Arvidsson, P.I.; Maguire, G.E.M.; Kruger, H.G.; Naicker, T. Enantioselective Organocatalyzed Transformations of β-Ketoesters. Chem. Rev. 2016, 116, 9375–9437. [Google Scholar] [CrossRef] [PubMed]
- Piovesana, S.; Schietroma, D.M.S.; Tulli, L.G.; Monaco, M.R.; Bella, M. Unsaturated β-ketoesters as versatile electrophiles in organocatalysis. Chem. Commun. 2010, 46, 5160–5162. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Tsutsumi, T.; Nakata, K.; Nakatsuji, H.; Tanabe, Y. Asymmetric Total Syntheses of Two 3-Acyl-5,6-dihydro-2H-pyrones: (R)-Podoblastin-S and (R)-Lachnelluloic Acid with Verification of the Absolute Configuration of (−)-Lachnelluloic Acid. Molecules 2017, 22, 69. [Google Scholar] [CrossRef] [PubMed]
- Song, C.E. An overview of cinchona alkaloids in chemistry. In Cinchona Alkaloids in Synthesis and Catalysis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; pp. 1–10. [Google Scholar] [CrossRef]
- Boratyński, P.J.; Zielińska-Błajet, M.; Skarżewski, J. Chapter Two—Cinchona Alkaloids—Derivatives and Applications. Alkaloids Chem. Bio. 2019, 82, 29–145. [Google Scholar] [CrossRef]
- Dijkstra, G.D.H.; Kellogg, R.M.; Wynberg, H. Conformational analysis of some chiral catalysts of the cinchona and ephedra family. The alkaloid catalyzed addition of aromatic thiols to cyclic α,β-unsaturated ketones. Red. Trav. Chim. Pays-Bas 1989, 108, 195–204. [Google Scholar] [CrossRef]
- Dijkstra, G.D.H.; Kellogg, R.M.; Wynberg, H.; Svendsen, J.S.; Marko, I.; Sharpless, K.B. Conformational study of cinchona alkaloids. A combined NMR, molecular mechanics and X-ray approach. J. Am. Chem. Soc. 1989, 111, 8069–8076. [Google Scholar] [CrossRef]
- Bürgi, T.; Baiker, A. Conformational Behavior of Cinchonidine in Different Solvents: A Combined NMR and ab Initio Investigation. J. Am. Chem. Soc. 1998, 120, 12920–12926. [Google Scholar] [CrossRef]
- Melchiorre, P. Cinchona-based Primary Amine Catalysis in the Asymmetric Functionalization of Carbonyl Compounds. Angew. Chem. Int. Ed. 2012, 51, 9748–9770. [Google Scholar] [CrossRef]
- Evans, G.J.S.; White, K.; Platts, J.A.; Tomkinson, N.C.O. Computational study of iminium ion formation: Effects of amine structure. Org. Biomol. Chem. 2006, 4, 2616–2627. [Google Scholar] [CrossRef]
- Tian, X.; Cassani, C.; Liu, Y.; Moran, A.; Urakawa, A.; Galzerano, P.; Arceo, E.; Melchiorre, P. Diastereodivergent Asymmetric Sulfa-Michael Additions of α-Branched Enones using a Single Chiral Organic Catalyst. J. Am. Chem. Soc. 2011, 133, 17934–17941. [Google Scholar] [CrossRef]
- Marigo, M.; Franzén, J.; Poulsen, T.B.; Zhuang, W.; Jørgensen, K.A. Asymmetric Organocatalytic Epoxidation of α,β-Unsaturated Aldehydes with Hydrogen Peroxide. J. Am. Chem. Soc. 2005, 127, 6964–6965. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, H.; O’Sullivan, T.P. Preparation of γ,δ-unsaturated-β-ketoesters: Lewis acid-catalysed CH insertion of ethyl diazoacetate into α,β-unsaturated aldehydes. Tetrahedron Lett. 2017, 58, 3533–3535. [Google Scholar] [CrossRef]
- Pietrusiewicz, K.M.; Monkiewicz, J. Anionic activation of stabilized ylides. A highly Z-stereoselective wittig reaction of (3-ethoxycarbonyl-2-oxopropylidene)triphenyl-phosphorane with aliphatic aldehydes. Tetrahedron Lett. 1986, 27, 739–742. [Google Scholar] [CrossRef]
- Porter, N.A.; Mitchell, J.C. Intramolecular alkylation of peroxides and hydroperoxides; peroxide transfer via peroxonium intermediates. Tetrahedron Lett. 1983, 24, 543–546. [Google Scholar] [CrossRef]
- Khazdooz, L.; Zarei, A.; Aghaei, H.; Azizi, G.; Gheisari, M.M. An efficient and selective method for the iodination and bromination of alcohols under mild conditions. Tetrahedron Lett. 2016, 57, 168–171. [Google Scholar] [CrossRef]
- Nguyen, T.L.; Ferrié, L.; Figadère, B. Synthesis of 3,5-disubstituted-1,2-dioxolanes: Access to analogues of mycangimycin and some rearrangement products. Tetrahedron Lett. 2016, 57, 5286–5289. [Google Scholar] [CrossRef]
- Pinet, A.; Nguyen, L.T.; Figadère, B.; Ferrié, L. Synthesis of 3,5-Disubstituted 1,2-Dioxolanes. Eur. J. Org. Chem. 2020, 2020, 7407–7416. [Google Scholar] [CrossRef]
- Pinet, A.; Figadère, B.; Ferrié, L. Access to Functionalized 3,5-Disubstituted 1,2-Dioxolanes under Mild Conditions through Indium(III) Chloride/Trimethylsilyl Chloride or Scandium(III) Triflate Catalysis. Adv. Synth. Catal. 2020, 362, 1190–1194. [Google Scholar] [CrossRef]
- Pinet, A.; Cojean, S.; Nguyen, L.T.; Vásquez-Ocmín, P.; Maciuk, A.; Loiseau, P.M.; Le Pape, P.; Figadère, B.; Ferrié, L. Anti-protozoal and anti-fungal evaluation of 3,5-disubstituted 1,2-dioxolanes. Bioorganic Med. Chem. Lett. 2021, 47, 128196. [Google Scholar] [CrossRef]
- Khazaei, A.; Rad, M.N.S.; Borazjani, M.K.; Saednia, S.; Borazjani, M.K.; Golbaghi, M.; Behrouz, S. Highly Efficient Etherification and Oxidation of Aromatic Alcohols Using Supported and Unsupported Phosphorus Pentoxide as a Heterogeneous Reagent. Synth. Commun. 2011, 41, 1544–1553. [Google Scholar] [CrossRef]
- Xu, W.; Wang, X.-B.; Wang, Z.-M.; Wu, J.-J.; Li, F.; Wang, J.; Kong, L.-Y. Synthesis and evaluation of donepezil–ferulic acid hybrids as multi-target-directed ligands against Alzheimer’s disease. MedChemComm 2016, 7, 990–998. [Google Scholar] [CrossRef]
- Vakulya, B.; Varga, S.; Csámpai, A.; Soós, T. Highly Enantioselective Conjugate Addition of Nitromethane to Chalcones Using Bifunctional Cinchona Organocatalysts. Org. Lett. 2005, 7, 1967–1969. [Google Scholar] [CrossRef] [PubMed]
- Cassani, C.; Martín-Rapún, R.; Arceo, E.; Bravo, F.; Melchiorre, P. Synthesis of 9-amino(9-deoxy)epi cinchona alkaloids, general chiral organocatalysts for the stereoselective functionalization of carbonyl compounds. Nat. Protoc. 2013, 8, 325–344. [Google Scholar] [CrossRef] [PubMed]
- Deng, R.; Han, T.-J.; Gao, X.; Yang, Y.-F.; Mei, G.-J. Further developments of β,γ-unsaturated α-ketoesters as versatile synthons in asymmetric catalysis. iScience 2022, 25, 103913. [Google Scholar] [CrossRef] [PubMed]
- Nagy, S.; Fehér, Z.; Dargó, G.; Barabás, J.; Garádi, Z.; Mátravölgyi, B.; Kisszékelyi, P.; Dargó, G.; Huszthy, P.; Höltzl, T.; et al. Comparison of Cinchona Catalysts Containing Ethyl or Vinyl or Ethynyl Group at Their Quinuclidine Ring. Materials 2019, 12, 3034. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Entry | Solvent | Conversion [a] | Isolated Yield | e.r. |
| 1 | MeCN | 40% | 26% | 69:31 |
| 2 | THF | 50% | 31% | 92:8 |
| 3 | EtOAc | 64% | 33% | 92:8 |
| 4 | Tol | 73% | 42% | 95:5 |
| 5 | EtOH | n.d. [b] | 13% (19%) [c] | 79:21 |
| 6 | H2O | 54% | 45% | 88:12 |
| 7 | CH2Cl2 | 75% | 52% | 93:7 |
| 8 | 1,2-DCE | 87% | 69% | 93:7 |
| 9 | CHCl3 | 73% | 49% | 96:4 |
| 10 | CCl4 | 61% | 42% | 94.5:5.5 |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Co-Catalyst | pKa | Temp. | Conversion [a] | Isolated Yield | e.r. |
| 1 | Triflic acid | −14.7 | r.t. | 60% | 41% | 57:43 |
| 2 | p-Toluenesulfonic acid | −2.8 | r.t. | 15% | 10% | 94:6 |
| 3 | Methanesulfonic acid | −1.9 | r.t. | 23% | 18% | 95.5:4.5 |
| 4 | Heptafluorobutyric acid | 0.4 | r.t. | 84% | 63% | 96:4 |
| 5 | Trifluoroacetic acid | 0.5 | r.t. | 77% | 58% | 96:4 |
| 6 | Pentafluorobenzoic acid | 1.5 | r.t. | 87% | 69% | 93:7 |
| 7 | Pentafluorobenzoic acid | 1.5 | 4 °C | 76% | 54% | 89:11 |
| 8 | Pentafluorobenzoic acid | 1.5 | 50 °C | 32% | 21% | 94:6 |
| 9 | Chloroacetic acid | 2.9 | r.t. | 25% | 17% | 86.5:13.5 |
| 10 | Tartaric acid | 2.9 | r.t. | 4% | n.d. | n.d. |
| 11 | 2,4-Bis(trifluoromethyl)benzoic acid | 3.3 | r.t. | 42% | 29% | 86:14 |
| 12 | 4-(Trifluoromethyl)benzoic acid | 3.6 | r.t. | 22% | 18% | 81:19 |
| 13 | Boc-L-phenylglycine | 3.9 | r.t. | 22% | 18% | 88:12 |
| 14 | Boc-D-phenylglycine | 3.9 | r.t. | 25% | 20% | 87:13 |
| 15 | Benzoic acid | 4.2 | r.t. | 8% | n.d. | n.d. |
 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | R | β-Keto Ester | Conversion [a] | Peroxide | Isolated Yield | e.r. | MW (g/mol) | Alcohol | Isolated Yield | MW (g/mol) |
| 1 | CH3 | 7 | 86% | 33 | 79% | 91:9 | 246.30 | 58 | 87% | 174.20 |
| 2 | CH3CH2 | 5 | 87% | 6 | 69% | 93:7 | 260.33 | 59 | 71% | 188.22 |
| 3 | CH3(CH2)2 | 8 | 74% | 34 | 52% | 93:7 | 274.36 | 60 | 77% | 202.25 |
| 4 | CH3(CH2)3 | 9 | 46% | 35 | 34% | 95:5 | 288.38 | 61 | 93% | 216.28 |
| 5 | CH3(CH2)4 | 10 | 50% | 36 | 43% | 95:5 | 302.41 | 62 | 93% | 230.30 |
| 6 | (CH3)2CH | 11 | n.d. | 37 | 27% | 90:10 | 274.36 | 63 | 93% | 202.25 |
| 7 | (CH3)2CHCH2 | 12 | 33% | 38 | 30% | 92:8 | 288.39 | 64 | 97% | 216.28 |
| 8 | Cy | 13 | 42% | 39 | 35% | 92:8 | 314.42 | 65 | 91% | 242.32 |
| 9 | C6H5 | 14 | 51% | 40 | 49% | 74:26 | 308.37 | 66 | 83% | 236.27 |
| 10 | 2-Naphthyl | 15 | 24% | 41 | 17% | 67:33 | 358.43 | 67 | 57% | 286.33 |
| 11 | C6H5CH2CH2 | 16 | 54% | 42 | 47% | 92:8 | 336.43 | 68 | 74% | 264.32 |
| 12 | 2-FC6H4 | 17 | 25% | 43 | 22% | 83:17 | 326.36 | 69 | 80% | 254.26 |
| 13 | 2-ClC6H4 | 18 | 29% | 44 | 25% | 91:9 | 342.82 | 70 | 84% | 270.71 |
| 14 | 2-BrC6H4 | 19 | 32% | 45 | 29% | 92:8 | 387.27 | 71 | 97% | 315.16 |
| 15 | 2-IC6H4 | 20 | 29% | 46 | 20% | 93:7 | 434.27 | 72 | 34% | 362.16 |
| 16 | 3-FC6H4 | 21 | 37% | 47 | 26% | 78:22 | 326.36 | 73 | 89% | 254.26 |
| 17 | 3-ClC6H4 | 22 | 42% | 48 | 17% | 78:22 | 342.82 | 74 | 86% | 270.71 |
| 18 | 3-BrC6H4 | 23 | 35% | 49 | 28% | 80:20 | 387.27 | 75 | 83% | 315.16 |
| 19 | 4-FC6H4 | 24 | 29% | 50 | 28% | 73:27 | 326.36 | 76 | 83% | 254.26 |
| 20 | 4-ClC6H4 | 25 | 26% | 51 | 24% | 76:24 | 342.82 | 77 | 80% | 270.71 |
| 21 | 4-BrC6H4 | 26 | 34% | 52 | 18% | 78:22 | 387.27 | 78 | 90% | 315.16 |
| 22 | 4-CF3C6H4 | 27 | 38% | 53 | 31% | 67:33 | 376.37 | 79 | 94% | 304.27 |
| 23 | 4-MeOC6H4 | 28 | 43% | 54 | 38% | 57:43 | 338.40 | 80 | 74% | 266.30 |
| 24 | BnOCH2 | 29 | 27% | 55 | 26% | 82:18 | 352.43 | 81 | 61% | 280.32 |
| 25 |  | 30 | 25% | 56 | 22% | n.d [b] | 352.38 | 82 | 94% | 280.28 |
| 26 | 2-Furyl | 31 | 7% | 57 | 6% | n.d [b] | 298.34 | - | - | - |
| 27 |  | 32 | 0% | - | 0% | - | - | - | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hennessy, M.C.; Gandhi, H.; O’Sullivan, T.P. Organocatalytic Asymmetric Peroxidation of γ,δ-Unsaturated β-Keto Esters—A Novel Route to Chiral Cycloperoxides. Molecules 2023, 28, 4317. https://doi.org/10.3390/molecules28114317
Hennessy MC, Gandhi H, O’Sullivan TP. Organocatalytic Asymmetric Peroxidation of γ,δ-Unsaturated β-Keto Esters—A Novel Route to Chiral Cycloperoxides. Molecules. 2023; 28(11):4317. https://doi.org/10.3390/molecules28114317
Chicago/Turabian StyleHennessy, Mary C., Hirenkumar Gandhi, and Timothy P. O’Sullivan. 2023. "Organocatalytic Asymmetric Peroxidation of γ,δ-Unsaturated β-Keto Esters—A Novel Route to Chiral Cycloperoxides" Molecules 28, no. 11: 4317. https://doi.org/10.3390/molecules28114317
APA StyleHennessy, M. C., Gandhi, H., & O’Sullivan, T. P. (2023). Organocatalytic Asymmetric Peroxidation of γ,δ-Unsaturated β-Keto Esters—A Novel Route to Chiral Cycloperoxides. Molecules, 28(11), 4317. https://doi.org/10.3390/molecules28114317