


Theoretical Description of Attosecond X-ray Absorption Spectroscopy of Frenkel Exciton Dynamics

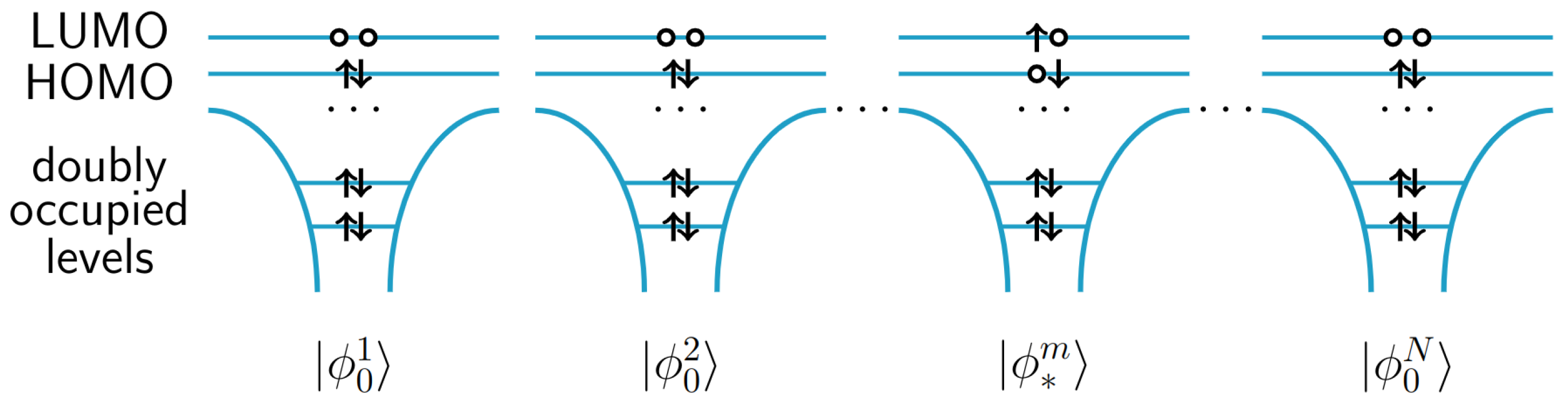{kind=link}
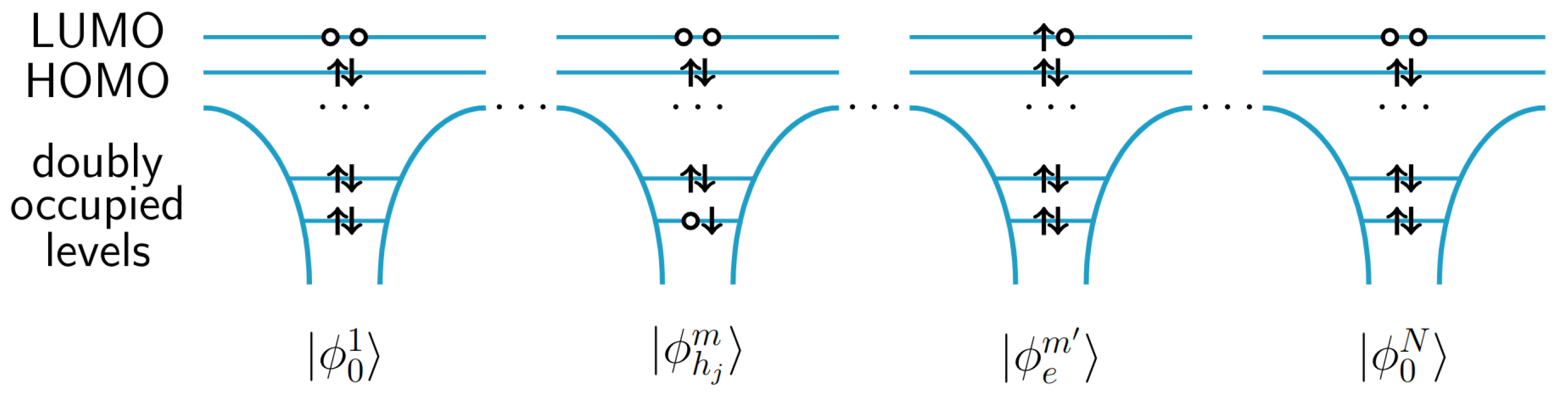{kind=link}
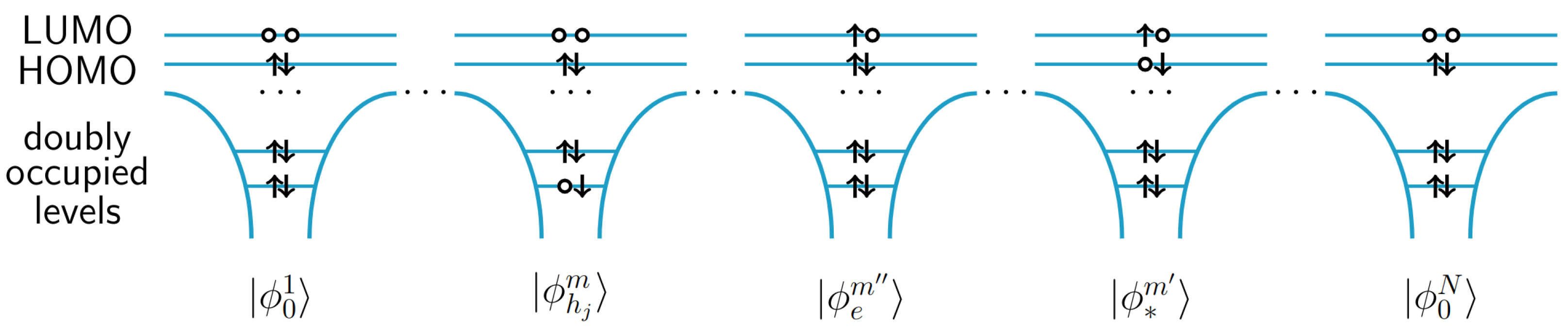{kind=link}
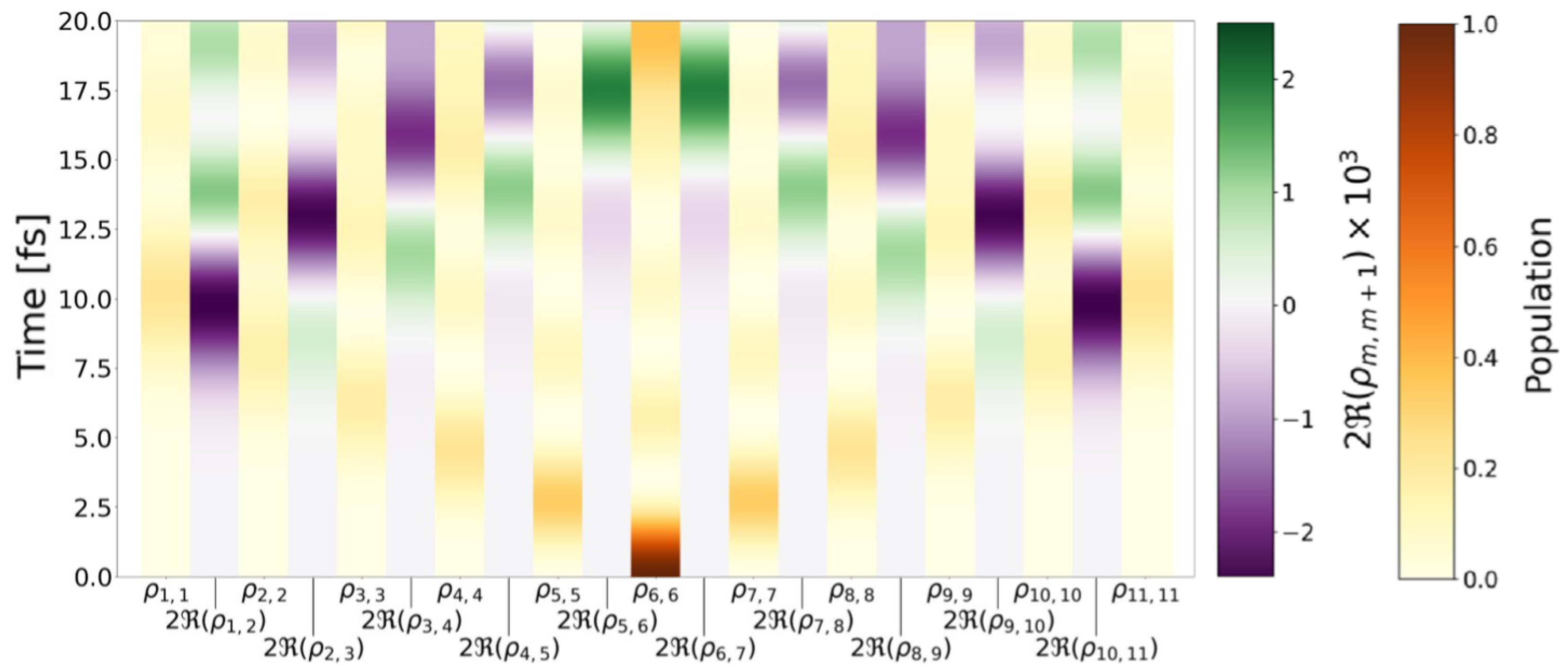{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Frenkel Exciton Dynamics
2.2. Interaction with an Ultrashort X-ray Probe Pulse
3. Results
3.1. X-ray Transitions below Fermi Level
3.2. X-ray Transitions above Fermi Level
3.3. Connection of the Time-Resolved X-ray Absorption cross Section to Frenkel Exciton Dynamics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| HOMO | Highest occupied molecular orbital |
| LUMO | Lowest unoccupied molecular orbital |
| Amplitude of the electric field of a probe pulse | |
| Pump-probe time delay | |
| Central frequency of a probe pulse | |
| Wave vector of a probe pulse | |
| Polarization vector of a probe pulse | |
| Probe pulse duration | |
| Index for the initially excited molecule | |
| j | Index for the atom j |
| m | Index for the molecule m |
| U | Coulomb energy of an electron-hole pair localized on a single site |
| V | Coupling between nearest-neighbor molecules |
| Degree of the exciton intersite delocalization | |
| Expansion coefficients of the representation of the time-dependent wave function as eigenstates | |
| Expansion coefficients of the representation of the eigenstates of Frenkel Hamiltonian as basis states | |
| Expansion coefficients of the solution of the time-dependent Schrödinger equation | |
| Transition matrix element between core electron orbital of the atom j of molecule m and | |
| HOMO of the same molecule | |
| Transition matrix element between core electron orbital of the atom j of the molecule m and | |
| HOMO of the next-neighbor molecules | |
| Transition matrix element between core electron orbital of the atom j of the molecule m and | |
| LUMO of the same molecule | |
| Transition matrix element between core electron of the atom j of the molecule m and | |
| LUMO of the next-neighbor molecules | |
| Ground state of the molecule m | |
| Ground state of a chain of molecules | |
| State which corresponds to an electron-hole pair located at the site m | |
| Basis state of Frenkel Hamiltonian | |
| Eigenstate of Frenkel Hamiltonian | |
| State of the molecule m with core hole at atom j | |
| State of the molecule m with one electron in LUMO | |
| State of the molecule m with hole in HOMO | |
| Final state after absorption | |
| Final state with a core hole at the atom j of the molecule m and an electron in LUMO of the molecule | |
| Final state with a core hole at the atom j of the molecule m, an electron-hole pair at the site and electron in LUMO at the site |
Appendix A
Appendix A.1. Derivation of Time-Resolved X-ray Absorption cross Section
Appendix A.2. Representation of the X-ray Absorption cross Section via Basis States
Appendix A.3. Derivation of X-ray Absorption cross Section for Transitions below Fermi Level
Appendix A.4. Derivation of X-ray Absorption cross Section for Transitions above Fermi Level
References
- Cheng, Y.C.; Fleming, G.R. Dynamics of Light Harvesting in Photosynthesis. Annu. Rev. Phys. Chem. 2009, 60, 241–262. [Google Scholar] [CrossRef] [PubMed]
- Dimitriev, O.P. Dynamics of Excitons in Conjugated Molecules and Organic Semiconductor Systems. Chem. Rev. 2022, 122, 8487–8593. [Google Scholar] [CrossRef] [PubMed]
- Duan, H.G.; Prokhorenko, V.I.; Cogdell, R.J.; Ashraf, K.; Stevens, A.L.; Thorwart, M.; Miller, D. Nature does not rely on long-lived electronic quantum coherence for photosynthetic energy transfer. Proc. Natl. Acad. Sci. USA 2017, 114, 8493–8498. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.Y.; Yan, Y.J.; Wei, J.H. Theoretical study on the exciton dynamics of coherent excitation energy transfer in the phycoerythrin 545 light-harvesting complex. Chin. Phys. B 2021, 31, 018201. [Google Scholar] [CrossRef]
- Wong, C.Y.; Alvey, R.M.; Turner, D.B.; Wilk, K.E.; Bryant, D.A.; Curmi, P.M.; Silbey, R.J.; Scholes, G.D. Electronic coherence lineshapes reveal hidden excitonic correlations in photosynthetic light harvesting. Nat. Chem. 2012, 4, 396–404. [Google Scholar] [CrossRef]
- Wang, L.; Allodi, M.A.; Engel, G.S. Quantum coherences reveal excited-state dynamics in biophysical systems. Nat. Rev. Chem. 2019, 3, 477–490. [Google Scholar] [CrossRef]
- Engel, G.S.; Calhoun, T.R.; Read, E.L.; Ahn, T.K.; Mančal, T.; Cheng, Y.C.; Blankenship, R.E.; Fleming, G.R. Evidence for wavelike energy transfer through quantum coherence in photosynthetic systems. Nature 2007, 446, 782–786. [Google Scholar] [CrossRef]
- Lambert, N.; Chen, Y.N.; Cheng, Y.C.; Li, C.M.; Chen, G.Y.; Nori, F. Quantum biology. Nat. Phys. 2013, 9, 10–18. [Google Scholar] [CrossRef]
- Fassioli, F.; Dinshaw, R.; Arpin, P.C.; Scholes, G.D. Photosynthetic light harvesting: Excitons and coherence. J. R. Soc. Interface 2014, 11, 20130901. [Google Scholar] [CrossRef]
- Cassette, E.; Pensack, R.D.; Mahler, B.; Scholes, G.D. Room-temperature exciton coherence and dephasing in two-dimensional nanostructures. Nat. Commun. 2015, 6, 6086. [Google Scholar] [CrossRef]
- Cao, J.; Cogdell, R.J.; Coker, D.F.; Duan, H.G.; Hauer, J.; Kleinekathöfer, U.; Jansen, T.L.C.; Mancal, T.; Miller, R.J.D.; Ogilvie, J.P.; et al. Quantum biology revisited. Sci. Adv. 2020, 6, eaaz4888. [Google Scholar] [CrossRef]
- Fassioli, F.; Olaya-Castro, A.; Scholes, G.D. Coherent Energy Transfer under Incoherent Light Conditions. J. Phys. Chem. Lett. 2012, 3, 3136–3142. [Google Scholar] [CrossRef] [PubMed]
- Rossi, G.M.; Mainz, R.E.; Yang, Y.; Scheiba, F.; Silva-Toledo, M.A.; Chia, S.H.; Keathley, P.D.; Fang, S.; Mücke, O.D.; Manzoni, C.; et al. Sub-cycle millijoule-level parametric waveform synthesizer for attosecond science. Nat. Photonics 2020, 14, 629–635. [Google Scholar] [CrossRef]
- Duris, J.; Li, S.; Driver, T.; Champenois, E.G.; MacArthur, J.P.; Lutman, A.A.; Zhang, Z.; Rosenberger, P.; Aldrich, J.W.; Coffee, R.; et al. Tunable isolated attosecond X-ray pulses with gigawatt peak power from a free-electron laser. Nat. Photonics 2020, 14, 30–36. [Google Scholar] [CrossRef]
- Buades, B.; Picón, A.; Berger, E.; León, I.; Di Palo, N.; Cousin, S.L.; Cocchi, C.; Pellegrin, E.; Martin, J.H.; Manas-Valero, S.; et al. Attosecond state-resolved carrier motion in quantum materials probed by soft X-ray XANES. Appl. Phys. Rev. 2021, 8, 011408. [Google Scholar] [CrossRef]
- Garratt, D.; Misiekis, L.; Wood, D.; Larsen, E.W.; Matthews, M.; Alexander, O.; Ye, P.; Jarosch, S.; Ferchaud, C.; Strüber, C.; et al. Direct observation of ultrafast exciton localization in an organic semiconductor with soft X-ray transient absorption spectroscopy. Nat. Commun. 2022, 13, 3414. [Google Scholar] [CrossRef] [PubMed]
- Zinchenko, K.S.; Ardana-Lamas, F.; Seidu, I.; Neville, S.P.; van der Veen, J.; Lanfaloni, V.U.; Schuurman, M.S.; Wörner, H.J. Sub-7-femtosecond conical-intersection dynamics probed at the carbon K-edge. Science 2021, 371, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Géneaux, R.; Kaplan, C.J.; Yue, L.; Ross, A.D.; Bækhøj, J.E.; Kraus, P.M.; Chang, H.T.; Guggenmos, A.; Huang, M.Y.; Zürch, M.; et al. Attosecond Time-Domain Measurement of Core-Level-Exciton Decay in Magnesium Oxide. Phys. Rev. Lett. 2020, 124, 207401. [Google Scholar] [CrossRef]
- Geneaux, R.; Marroux, H.J.B.; Guggenmos, A.; Neumark, D.M.; Leone, S.R. Transient absorption spectroscopy using high harmonic generation: A review of ultrafast X-ray dynamics in molecules and solids. Philos. Trans. A Math. Phys. Eng. Sci. 2019, 377, 20170463. [Google Scholar] [CrossRef]
- Krausz, F.; Ivanov, M. Attosecond physics. Rev. Mod. Phys. 2009, 81, 163–234. [Google Scholar] [CrossRef]
- Popova-Gorelova, D. Imaging Electron Dynamics with Ultrashort Light Pulses: A Theory Perspective. Appl. Sci. 2018, 8, 318. [Google Scholar] [CrossRef]
- Saes, M.; Bressler, C.; Abela, R.; Grolimund, D.; Johnson, S.L.; Heimann, P.A.; Chergui, M. Observing Photochemical Transients by Ultrafast X-ray Absorption Spectroscopy. Phys. Rev. Lett. 2003, 90, 047403. [Google Scholar] [CrossRef] [PubMed]
- Bhattacherjee, A.; Leone, S.R. Ultrafast X-ray Transient Absorption Spectroscopy of Gas-Phase Photochemical Reactions: A New Universal Probe of Photoinduced Molecular Dynamics. Accounts Chem. Res. 2018, 51, 3203–3211. [Google Scholar] [CrossRef] [PubMed]
- Khakhulin, D.; Otte, F.; Biednov, M.; Bömer, C.; Choi, T.K.; Diez, M.; Galler, A.; Jiang, Y.; Kubicek, K.; Lima, F.A.; et al. Ultrafast X-ray Photochemistry at European XFEL: Capabilities of the Femtosecond X-ray Experiments (FXE) Instrument. Appl. Sci. 2020, 10, 995. [Google Scholar] [CrossRef]
- Shakya, Y.; Inhester, L.; Arnold, C.; Welsch, R.; Santra, R. Ultrafast time-resolved X-ray absorption spectroscopy of ionized urea and its dimer through ab initio nonadiabatic dynamics. Struct. Dyn. 2021, 8, 034102. [Google Scholar] [CrossRef]
- Frenkel, J. On the Transformation of light into Heat in Solids. I. Phys. Rev. 1931, 37, 17–44. [Google Scholar] [CrossRef]
- Saikin, S.K.; Eisfeld, A.; Valleau, S.; Aspuru-Guzik, A. Photonics meets excitonics: Natural and artificial molecular aggregates. Nanophotonics 2013, 2, 21–38. [Google Scholar] [CrossRef]
- Bardeen, C.J. The Structure and Dynamics of Molecular Excitons. Annu. Rev. Phys. Chem. 2014, 65, 127–148. [Google Scholar] [CrossRef]
- Agranovich, V. Excitations in Organic Solids; Oxford University Press: Oxford, UK, 2008. [Google Scholar]
- Akselrod, G.M.; Deotare, P.B.; Thompson, N.J.; Lee, J.; Tisdale, W.A.; Baldo, M.A.; Menon, V.M.; Bulović, V. Visualization of exciton transport in ordered and disordered molecular solids. Nat. Commun. 2014, 5, 3646. [Google Scholar] [CrossRef]
- Wan, Y.; Guo, Z.; Zhu, T.; Yan, S.; Johnson, J.; Huang, L. Cooperative singlet and triplet exciton transport in tetracene crystals visualized by ultrafast microscopy. Nat. Chem. 2015, 7, 785–792. [Google Scholar] [CrossRef]
- Zhu, T.; Wan, Y.; Huang, L. Direct Imaging of Frenkel Exciton Transport by Ultrafast Microscopy. Accounts Chem. Res. 2017, 50, 1725–1733. [Google Scholar] [CrossRef] [PubMed]
- Stadtmüller, B.; Emmerich, S.; Jungkenn, D.; Haag, N.; Rollinger, M.; Eich, S.; Maniraj, M.; Aeschlimann, M.; Cinchetti, M.; Mathias, S. Strong modification of the transport level alignment in organic materials after optical excitation. Nat. Commun. 2019, 10, 1470. [Google Scholar] [CrossRef] [PubMed]
- Emmerich, S.; Hedwig, S.; Arnoldi, B.; Stöckl, J.; Haag, F.; Hemm, R.; Cinchetti, M.; Mathias, S.; Stadtmüller, B.; Aeschlimann, M. Ultrafast Charge-Transfer Exciton Dynamics in C60 Thin Films. J. Phys. Chem. C 2020, 124, 23579–23587. [Google Scholar] [CrossRef] [PubMed]
- Santra, R.; Yakovlev, V.S.; Pfeifer, T.; Loh, Z.H. Theory of attosecond transient absorption spectroscopy of strong-field-generated ions. Phys. Rev. A 2011, 83, 033405. [Google Scholar] [CrossRef]
- Santra, R. Concepts in X-ray physics. J. Phys. B At. Mol. Opt. Phys. 2008, 42, 023001. [Google Scholar] [CrossRef]
- Albert, V.V.; Badaeva, E.; Kilina, S.; Sykora, M.; Tretiak, S. The Frenkel exciton Hamiltonian for functionalized Ru(II)–bpy complexes. J. Lumin. 2011, 131, 1739–1746. [Google Scholar] [CrossRef]
- Farahvash, A.; Lee, C.K.; Sun, Q.; Shi, L.; Willard, A.P. Machine learning Frenkel Hamiltonian parameters to accelerate simulations of exciton dynamics. J. Chem. Phys. 2020, 153, 074111. [Google Scholar] [CrossRef]
- Nematiaram, T.; Padula, D.; Troisi, A. Bright Frenkel Excitons in Molecular Crystals: A Survey. Chem. Mater. 2021, 33, 3368–3378. [Google Scholar] [CrossRef]
- Scholes, G.D.; Smyth, C. Perspective: Detecting and measuring exciton delocalization in photosynthetic light harvesting. J. Chem. Phys. 2014, 140, 110901. [Google Scholar] [CrossRef]
- Chenu, A.; Scholes, G.D. Coherence in Energy Transfer and Photosynthesis. Annu. Rev. Phys. Chem. 2015, 66, 69–96. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hansen, T.; Bezriadina, T.; Popova-Gorelova, D. Theoretical Description of Attosecond X-ray Absorption Spectroscopy of Frenkel Exciton Dynamics. Molecules 2023, 28, 4502. https://doi.org/10.3390/molecules28114502
Hansen T, Bezriadina T, Popova-Gorelova D. Theoretical Description of Attosecond X-ray Absorption Spectroscopy of Frenkel Exciton Dynamics. Molecules. 2023; 28(11):4502. https://doi.org/10.3390/molecules28114502
Chicago/Turabian StyleHansen, Tim, Tatiana Bezriadina, and Daria Popova-Gorelova. 2023. "Theoretical Description of Attosecond X-ray Absorption Spectroscopy of Frenkel Exciton Dynamics" Molecules 28, no. 11: 4502. https://doi.org/10.3390/molecules28114502
APA StyleHansen, T., Bezriadina, T., & Popova-Gorelova, D. (2023). Theoretical Description of Attosecond X-ray Absorption Spectroscopy of Frenkel Exciton Dynamics. Molecules, 28(11), 4502. https://doi.org/10.3390/molecules28114502