
Electrode Potential-Dependent Studies of Protein Adsorption on Ti6Al4V Alloy


 and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Atomic Force Microscopy (AFM)
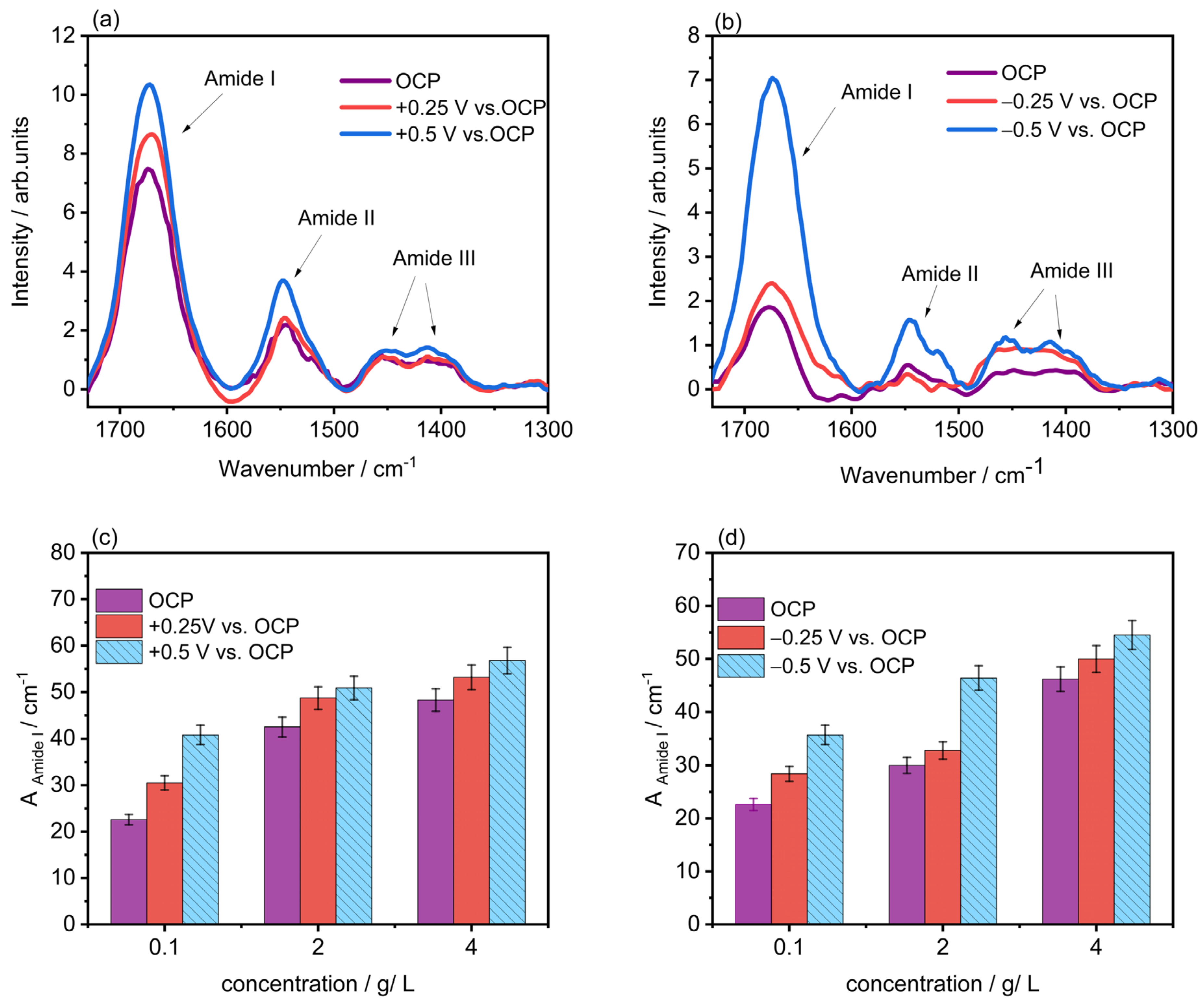
2.2. Polarization-Modulation Infrared Reflection-Absorption Spectroscopy (PM-IRRAS)
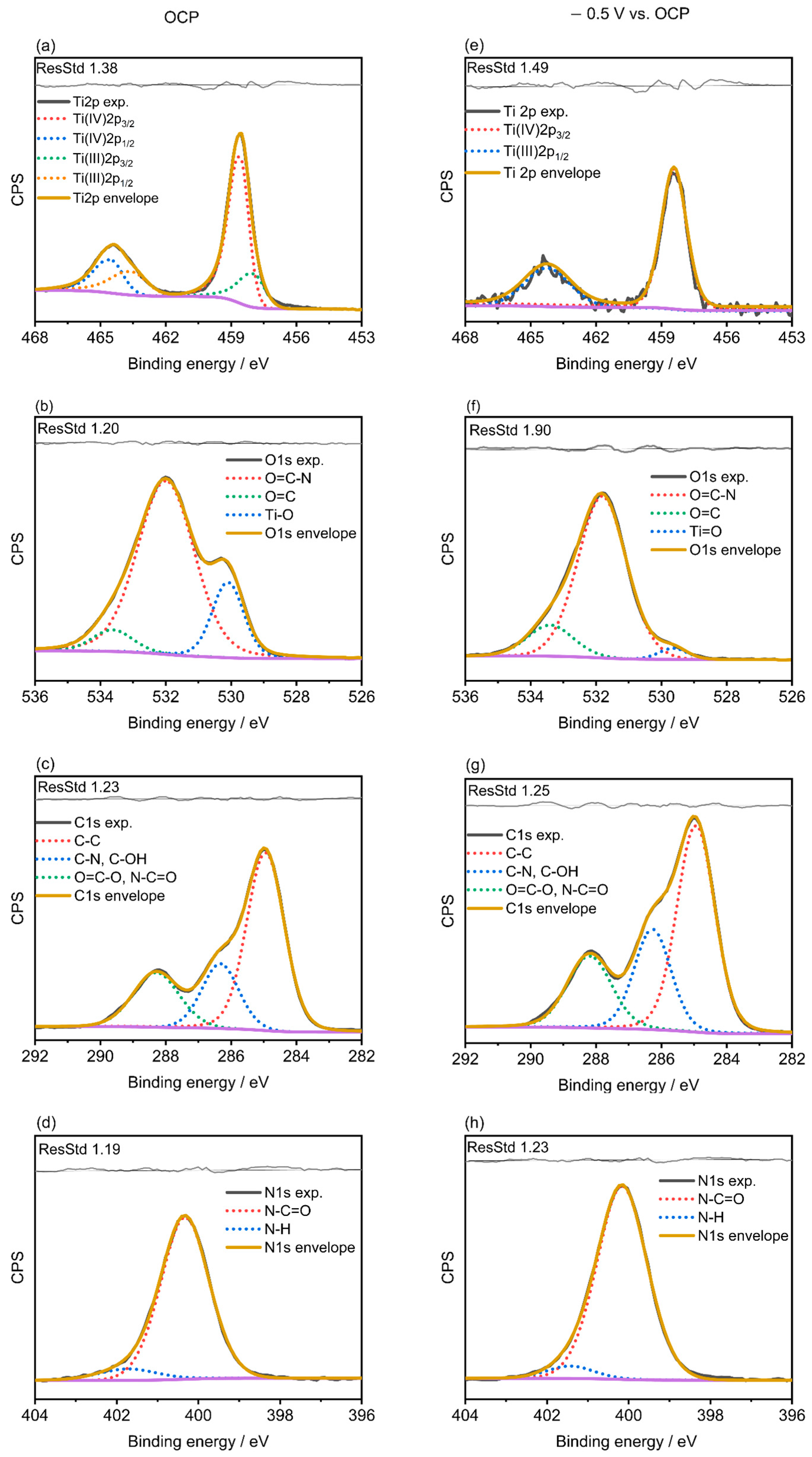
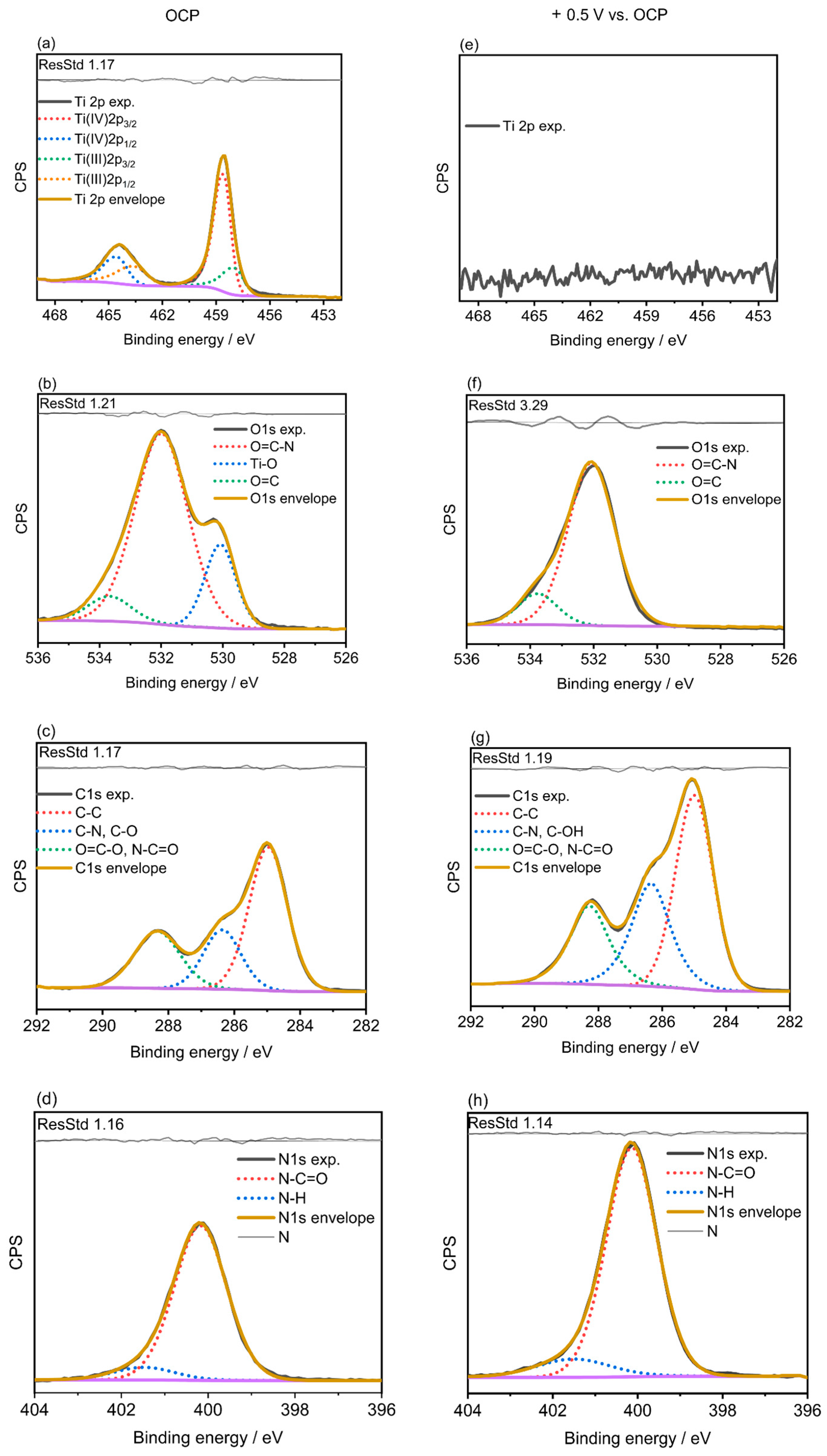
2.3. X-ray Photoelectron Spectroscopy (XPS)
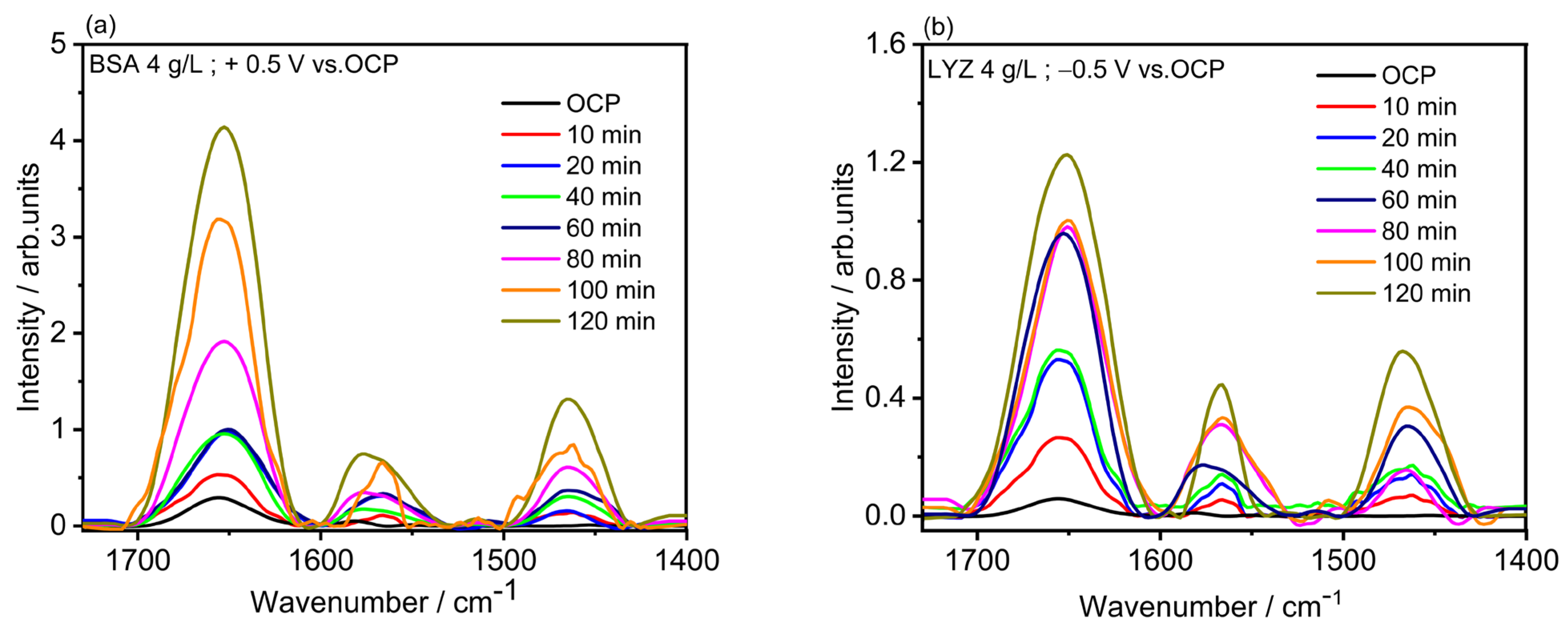
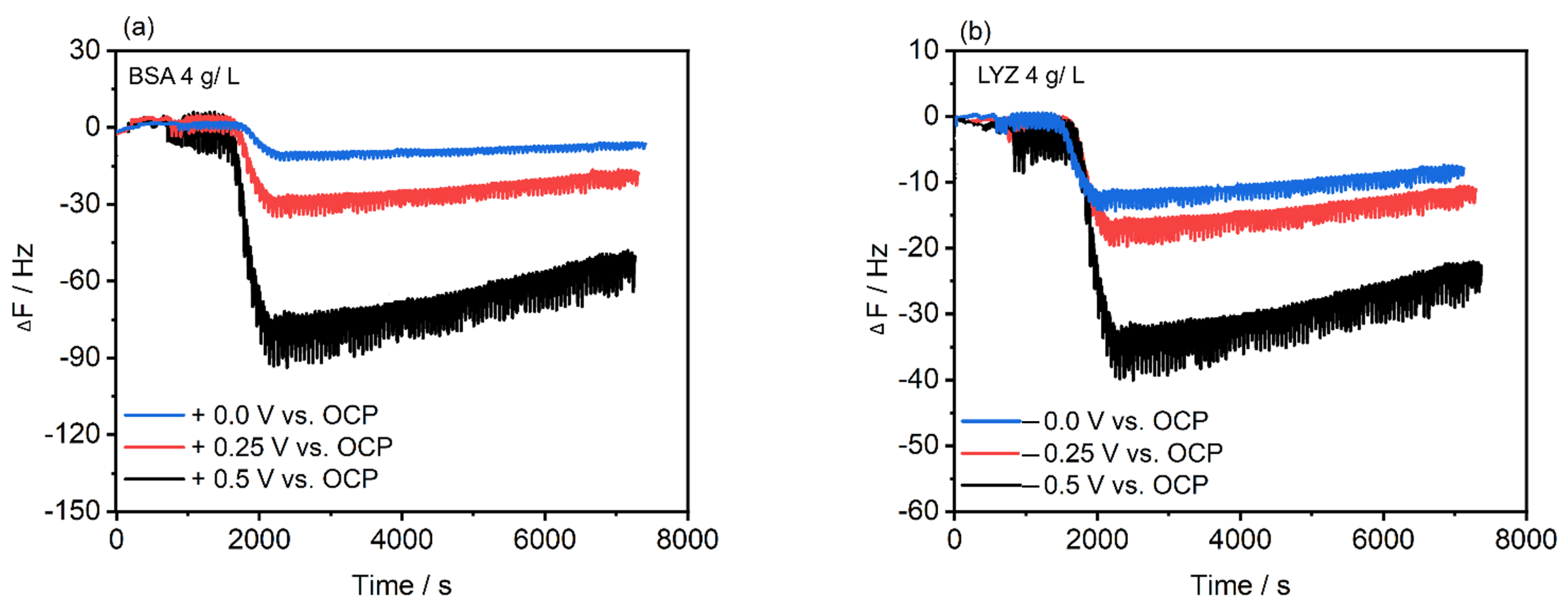
2.4. Electrochemical Polarization-Modulation Infrared Reflection-Absorption Spectroscopy (E-PM-IRRAS) and Electrochemical Quartz Crystal Microbalance (E-QCM)
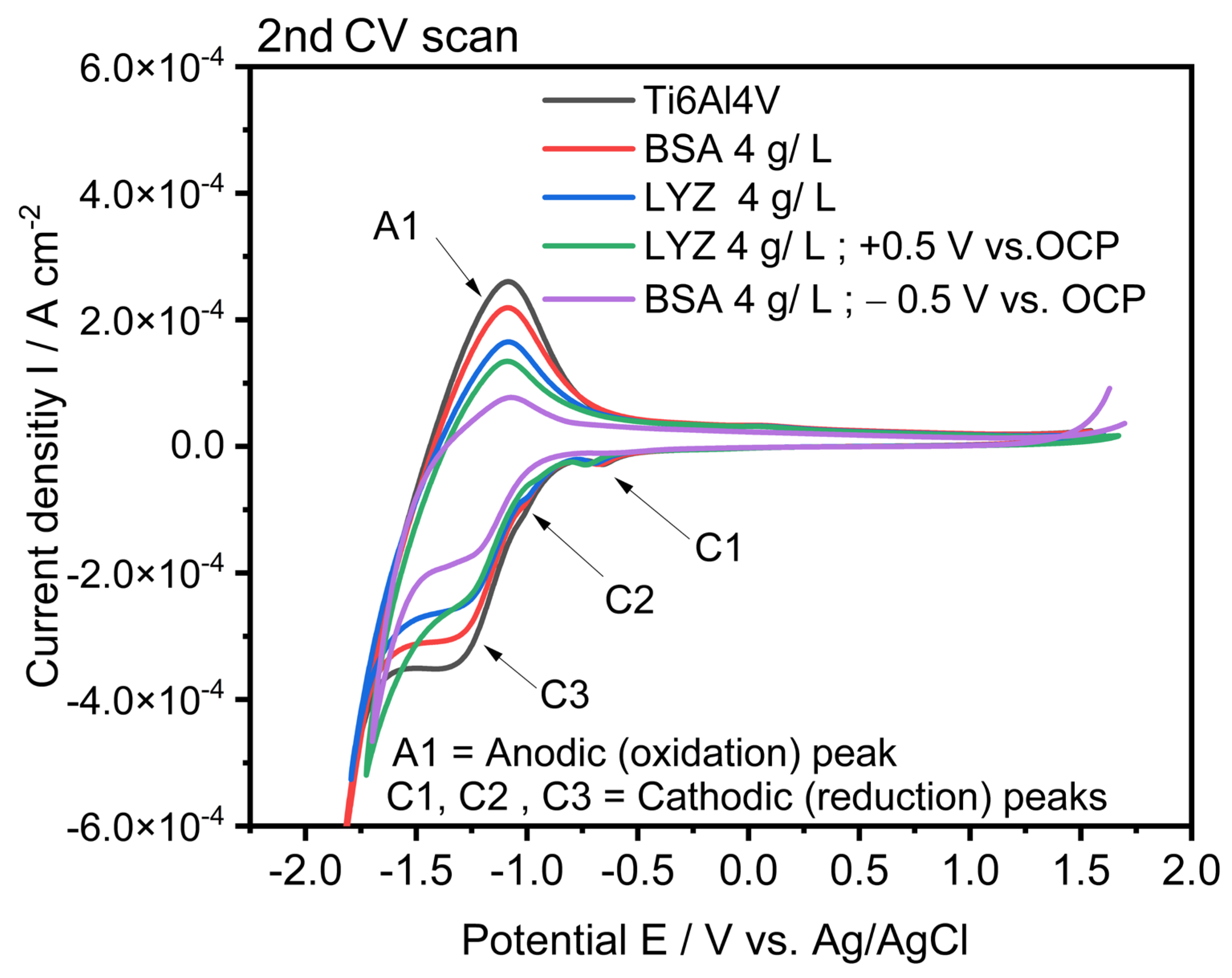
2.5. Electrochemical Characterization
3. Materials and Methods
3.1. Surface Pre-Conditioning and Electrochemical Modification of the Substrate Surfaces
3.2. Protein Adsorption
3.3. Atomic Force Microscopy (AFM)
3.4. X-ray Photoelectron Spectroscopy (XPS)
3.5. Polarization-Modulation Infrared Reflection-Absorption Spectroscopy (PM-IRRAS)
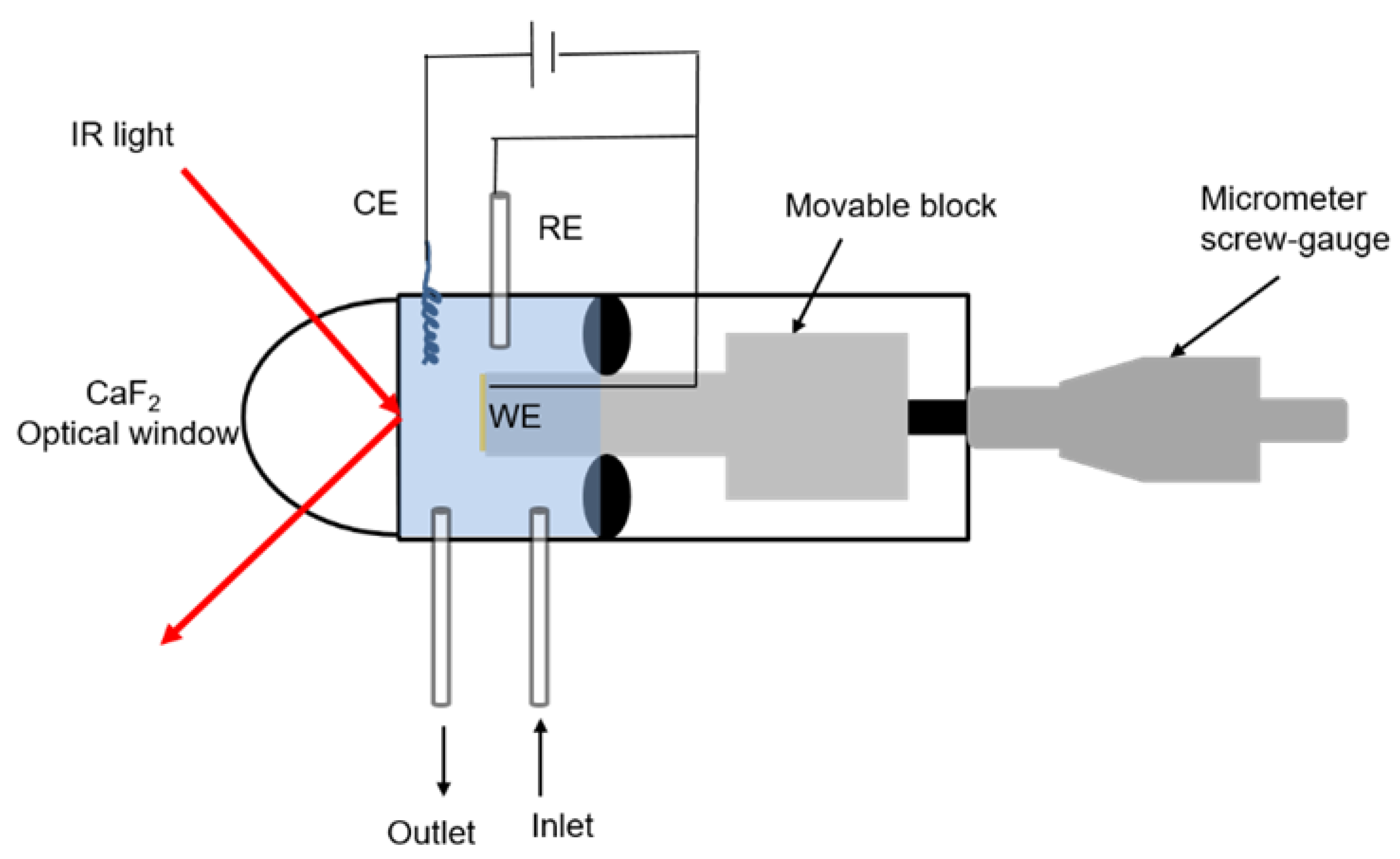
3.6. Electrochemical Polarization-Modulation Infrared Reflection-Absorption Spectroscopy (E-PM-IRRAS)
3.7. Electrochemical Quartz Crystal Microbalance (E-QCM)
3.8. Electrochemical Characterization
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Gray, J.J. The interaction of proteins with solid surfaces. Curr. Opin. Struct. Biol. 2004, 14, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Brunette, D.M.; Tengvall, P.; Textor, M.; Thomsen, P. Titanium in Medicine. In Material Science, Surface Science, Engineering, Biological Responses and Medical Applications; Springer: Berlin, Germany, 2001; pp. 145–162. [Google Scholar]
- Norde, W. The behavior of proteins at interfaces, with special attention to the role of the structure stability of the protein molecule. Clin. Mater. 1992, 11, 85–91. [Google Scholar] [CrossRef]
- Dee, K.C.; Puleo, D.A.; Bizios, R. An Introduction to Tissue-Biomaterial Interactions; Wiley-Liss: Hoboken, NJ, USA, 2002. [Google Scholar] [CrossRef]
- Sittig, C.; Textor, M.; Spencer, N.D.; Wieland, M.; Vallotton, P.-H. Surface characterization. J. Mater. Sci. Mater. Med. 1999, 10, 35–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shayganpour, A.; Rebaudi, A.; Cortella, P.; Diaspro, A.; Salerno, M. Electrochemical coating of dental implants with anodic porous titania for enhanced osteointegration. Beilstein J. Nanotechnol. 2015, 6, 2183–2192. [Google Scholar] [CrossRef] [PubMed]
- Birch, M.A.; Johnson-Lynn, S.; Nouraei, S.; Wu, Q.-B.; Ngalim, S.; Lu, W.-J.; Watchorn, C.; Yang, T.-Y.; McCaskie, A.W.; Roy, S. Effect of electrochemical structuring of Ti6Al4V on osteoblast behaviour in vitro. Biomed. Mater. 2012, 7, 035016. [Google Scholar] [CrossRef] [PubMed]
- Gao, A.; Hang, R.; Bai, L.; Tang, B.; Chu, P.K. Electrochemical surface engineering of titanium-based alloys for biomedical application. Electrochim. Acta 2018, 271, 699–718. [Google Scholar] [CrossRef]
- Metoki, N.; Liu, L.; Beilis, E.; Eliaz, N.; Mandler, D. Preparation and Characterization of Alkylphosphonic Acid Self-Assembled Monolayers on Titanium Alloy by Chemisorption and Electrochemical Deposition. Langmuir 2014, 30, 6791–6799. [Google Scholar] [CrossRef]
- Yamauchi, R.; Itabashi, T.; Wada, K.; Tanaka, T.; Kumagai, G.; Ishibashi, Y. Photofunctionalised Ti6Al4V implants enhance early phase osseointegration. Bone Jt. Res. 2017, 6, 331–336. [Google Scholar] [CrossRef]
- Madore, C.; Piotrowski, O.; Landolt, D. Through-Mask Electrochemical Micromachining of Titanium. J. Electrochem. Soc. 1999, 146, 2526–2532. [Google Scholar] [CrossRef]
- Beutner, R.; Michael, J.; Schwenzer, B.; Scharnweber, D. Biological nano-functionalization of titanium-based biomaterial surfaces: A flexible toolbox. J. R. Soc. Interface 2009, 7, S93–S105. [Google Scholar] [CrossRef] [Green Version]
- Kern, P.; Veh, J.; Michler, J. New developments in through-mask electrochemical micromachining of titanium. J. Micromech. Microeng. 2007, 17, 1168–1177. [Google Scholar] [CrossRef]
- Imamura, K.; Shimomura, M.; Nagai, S.; Akamatsu, M.; Nakanishi, K. Adsorption characteristics of various proteins to a titanium surface. J. Biosci. Bioeng. 2008, 106, 273–278. [Google Scholar] [CrossRef]
- Göhring, H.; Paulus, M.; Salmen, P.; Wirkert, F.; Kruse, T.; Degen, P.; Stuhr, S.; Rehage, H.; Tolan, M. Salt induced reduction of lysozyme adsorption at charged interfaces. J. Phys. Condens. Matter 2015, 27, 235103. [Google Scholar] [CrossRef]
- Silva, R.A.; Urzúa, M.D.; Petri, D.F.S.; Dubin, P.L. Protein Adsorption onto Polyelectrolyte Layers: Effects of Protein Hydrophobicity and Charge Anisotropy. Langmuir 2010, 26, 14032–14038. [Google Scholar] [CrossRef]
- Ei, H.E.; Nakama, Y.; Tanaka, H.; Imanaka, H.; Ishida, N.; Imamura, K. Adsorption of lysozyme on base metal surfaces in the presence of an external electric potential. Colloids Surf. B Biointerfaces 2016, 147, 9–16. [Google Scholar] [CrossRef]
- Beykal, B.; Herzberg, M.; Oren, Y.; Mauter, M.S. Influence of surface charge on the rate, extent, and structure of adsorbed Bovine Serum Albumin to gold electrodes. J. Colloid Interface Sci. 2015, 460, 321–328. [Google Scholar] [CrossRef]
- Brusatori, M.A.; Tie, Y.; Van Tassel, P.R. Protein Adsorption Kinetics under an Applied Electric Field: An Optical Waveguide Lightmode Spectroscopy Study. Langmuir 2003, 19, 5089–5097. [Google Scholar] [CrossRef]
- Ying, P.; Viana, A.S.; Abrantes, L.M.; Jin, G. Adsorption of human serum albumin onto gold: A combined electrochemical and ellipsometric study. J. Colloid Interface Sci. 2004, 279, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Moulton, S.; Barisci, J.; Bath, A.; Stella, R.; Wallace, G. Investigation of protein adsorption and electrochemical behavior at a gold electrode. J. Colloid Interface Sci. 2003, 261, 312–319. [Google Scholar] [CrossRef]
- Oliva, F.Y.; Cámara, O.R.; Avalle, L.B. Adsorption of human serum albumin on electrochemical titanium dioxide electrodes: Protein-oxide surface interaction effects studied by electrochemical techniques. J. Electroanal. Chem. 2009, 633, 19–34. [Google Scholar] [CrossRef]
- Engelhardt, K.; Rumpel, A.; Walter, J.; Dombrowski, J.; Kulozik, U.; Braunschweig, B.; Peukert, W. Protein Adsorption at the Electrified Air-Water Interface: Implications on Foam Stability. Langmuir 2012, 28, 7780–7787. [Google Scholar] [CrossRef] [PubMed]
- Calonder, C.; Tie, Y.; Van Tassel, P.R. History dependence of protein adsorption kinetics. Proc. Natl. Acad. Sci. USA 2001, 98, 10664–10669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Yang, S.; Kong, J.; Dong, A.; Yu, S. Obtaining information about protein secondary structures in aqueous solution using Fourier transform IR spectroscopy. Nat. Protoc. 2015, 10, 382–396. [Google Scholar] [CrossRef] [PubMed]
- Norde, W. Driving forces for protein adsorption at solid surfaces. Macromol. Symp. 1996, 103, 5–18. [Google Scholar] [CrossRef]
- Ouberai, M.M.; Xu, K.; Welland, M.E. Effect of the interplay between protein and surface on the properties of adsorbed protein layers. Biomaterials 2014, 35, 6157–6163. [Google Scholar] [CrossRef] [Green Version]
- Stuart, B.H. Infrared Spectroscopy: Fundamentals and Applications; John Wiley and Sons Ltd.: Chichester, UK, 2004; ISBN 0470854278. [Google Scholar]
- Shvab, R.; Hryha, E.; Nyborg, L. Surface chemistry of the titanium powder studied by XPS using internal standard reference. Powder Met. 2016, 60, 42–48. [Google Scholar] [CrossRef]
- Ajami, E.; Aguey-Zinsou, K.-F. Calcium Phosphate Growth at Electropolished Titanium Surfaces. J. Funct. Biomater. 2012, 3, 327–348. [Google Scholar] [CrossRef] [Green Version]
- Pradier, C.M.; Costa, D.; Rubio, C.; Compère, C.; Marcus, P. Role of salts on BSA adsorption on stainless steel in aqueous solutions. I. FT-IRRAS and XPS characterization: Bovine serum albumin adsorption on stainless steel. Surf. Interface Anal. 2002, 34, 50–54. [Google Scholar] [CrossRef]
- Muñoz, A.I.; Mischler, S. Interactive Effects of Albumin and Phosphate Ions on the Corrosion of CoCrMo Implant Alloy. J. Electrochem. Soc. 2007, 154, C562–C570. [Google Scholar] [CrossRef]
- Solak, E.K. Preparation and Characterization of IPN Microspheres for Controlled Delivery of Naproxen. JBNB 2011, 02, 445–453. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.; Gurau, M.; Kim, J.; Cremer, P.S. Investigations of Lysozyme Adsorption at the Air/Water and Quartz/Water Interfaces by Vibrational Sum Frequency Spectroscopy. Langmuir 2002, 18, 2807–2811. [Google Scholar] [CrossRef]
- Kim, J.; Somorjai, G.A. Molecular Packing of Lysozyme, Fibrinogen, and Bovine Serum Albumin on Hydrophilic and Hydrophobic Surfaces Studied by Infrared-Visible Sum Frequency Generation and Fluorescence Microscopy. J. Am. Chem. Soc. 2003, 125, 3150–3158. [Google Scholar] [CrossRef]
- Van der Veen, M.; Stuart, M.C.; Norde, W. Spreading of proteins and its effect on adsorption and desorption kinetics. Colloids Surf. B Biointerfaces 2007, 54, 136–142. [Google Scholar] [CrossRef]
- Larsericsdotter, H.; Oscarsson, S.; Buijs, J. Structure, stability, and orientation of BSA adsorbed to silica. J. Colloid Interface Sci. 2005, 289, 26–35. [Google Scholar] [CrossRef]
- Dixon, M.C. Quartz crystal microbalance with dissipation monitoring: Enabling real-time characterization of biological materials and their interactions. J. Biomol. Tech. 2008, 19, 151–158. [Google Scholar]
- Wei, T.; Kaewtathip, S.; Shing, K. Buffer Effect on Protein Adsorption at Liquid/Solid Interface. J. Phys. Chem. C 2009, 113, 2053–2062. [Google Scholar] [CrossRef]
- Peng, Z.; Hidajat, K.; Uddin, M. Conformational change of adsorbed and desorbed bovine serum albumin on nano-sized magnetic particles. Colloids Surf. B Biointerfaces 2004, 33, 15–21. [Google Scholar] [CrossRef]
- Vermonden, T.; Giacomelli, C.E.; Norde, W. Reversibility of Structural Rearrangements in Bovine Serum Albumin during Homomolecular Exchange from AgI Particles. Langmuir 2001, 17, 3734–3740. [Google Scholar] [CrossRef]
- Nejadnik, M.R.; Garcia, C.D. Staining proteins: A simple method to increase the sensitivity of ellipsometric measurements in adsorption studies. Colloids Surf. B Biointerfaces 2011, 82, 253–257. [Google Scholar] [CrossRef] [Green Version]
- Wena, J.; Arakawa, T. Refractive Index of Proteins in Aqueous Sodium Chloride. Anal. Biochem. 2000, 280, 327–329. [Google Scholar] [CrossRef]
- Siddiqui, M.A.; Ullah, I.; Liu, H.; Zhang, S.; Ren, L.; Yang, K. Preliminary study of adsorption behavior of bovine serum albumin (BSA) protein and its effect on antibacterial and corrosion property of Ti-3Cu alloy. J. Mater. Sci. Technol. 2020, 80, 117–127. [Google Scholar] [CrossRef]
- Makivić, N.; Cho, J.-Y.; Harris, K.D.; Tarascon, J.-M.; Limoges, B.; Balland, V. Evidence of Bulk Proton Insertion in Nanostructured Anatase and Amorphous TiO2 Electrodes. Chem. Mater. 2021, 33, 3436–3448. [Google Scholar] [CrossRef]
- Silva-Bermudez, P.; Rodil, S. An overview of protein adsorption on metal oxide coatings for biomedical implants. Surf. Coat. Technol. 2013, 233, 147–158. [Google Scholar] [CrossRef]
- Felgueiras, H.; Antunes, J.; Martins, M.; Barbosa, M. Fundamentals of protein and cell interactions in biomaterials. In Peptides and Proteins as Biomaterials for Tissue Regeneration and Repair; Elsevier: Amsterdam, The Netherlands, 2018; pp. 1–27. [Google Scholar] [CrossRef]
- Nečas, D.; Klapetek, P. Gwyddion: An open-source software for SPM data analysis. Open Phys. 2012, 10, 181–188. [Google Scholar] [CrossRef]
- Brand, I. Application of Polarization Modulation Infrared Reflection Absorption Spectrosocpy Under Electrochemical Control for Structural Studies of Biomimetic Assemblies. Z. Phys. Chem. 2015, 230, 133–183. [Google Scholar] [CrossRef]
- Fu, K.; Griebenow, K.; Hsieh, L.; Klibanov, A.M.; Langer, R. FTIR characterization of the secondary structure of proteins encapsulated within PLGA microspheres. J. Control. Release 1999, 58, 357–366. [Google Scholar] [CrossRef]
- Höök, F.; Kasemo, B.; Nylander, T.; Fant, C.; Sott, K.; Elwing, H. Variations in Coupled Water, Viscoelastic Properties, and Film Thickness of a Mefp-1 Protein Film during Adsorption and Cross-Linking: A Quartz Crystal Microbalance with Dissipation Monitoring, Ellipsometry, and Surface Plasmon Resonance Study. Anal. Chem. 2001, 73, 5796–5804. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Atomic Percentages (at.-%) | |||
|---|---|---|---|---|
| Ti2p | O1s | C1s | N1s | |
| LYZ at OCP | 3.8 | 38.7 | 48.3 | 7.4 |
| LYZ at −0.5 V vs. OCP | 2.2 | 25.4 | 56.2 | 12.7 |
| BSA at OCP | 2.9 | 26.9 | 61.1 | 8.9 |
| BSA at + 0.5 V vs. OCP | - | 18.5 | 67.8 | 13.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duderija, B.; González-Orive, A.; Ebbert, C.; Neßlinger, V.; Keller, A.; Grundmeier, G. Electrode Potential-Dependent Studies of Protein Adsorption on Ti6Al4V Alloy. Molecules 2023, 28, 5109. https://doi.org/10.3390/molecules28135109
Duderija B, González-Orive A, Ebbert C, Neßlinger V, Keller A, Grundmeier G. Electrode Potential-Dependent Studies of Protein Adsorption on Ti6Al4V Alloy. Molecules. 2023; 28(13):5109. https://doi.org/10.3390/molecules28135109
Chicago/Turabian StyleDuderija, Belma, Alejandro González-Orive, Christoph Ebbert, Vanessa Neßlinger, Adrian Keller, and Guido Grundmeier. 2023. "Electrode Potential-Dependent Studies of Protein Adsorption on Ti6Al4V Alloy" Molecules 28, no. 13: 5109. https://doi.org/10.3390/molecules28135109
APA StyleDuderija, B., González-Orive, A., Ebbert, C., Neßlinger, V., Keller, A., & Grundmeier, G. (2023). Electrode Potential-Dependent Studies of Protein Adsorption on Ti6Al4V Alloy. Molecules, 28(13), 5109. https://doi.org/10.3390/molecules28135109