
Investigation of the Origin of High Photoluminescence Quantum Yield in Thienyl-S,S-dioxide AIEgens Oligomers by Temperature Dependent Optical Spectroscopy


Abstract
:1. Introduction
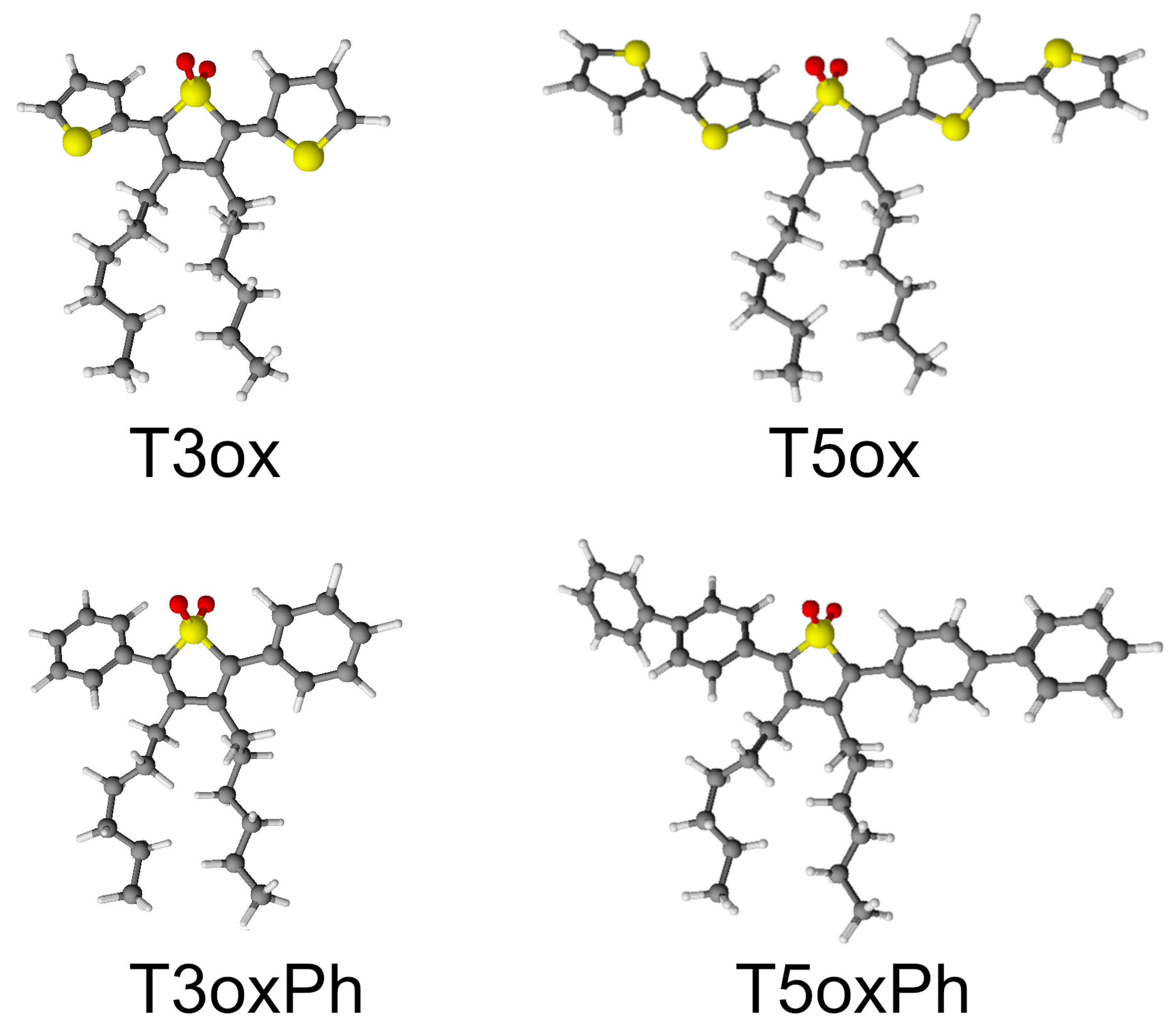
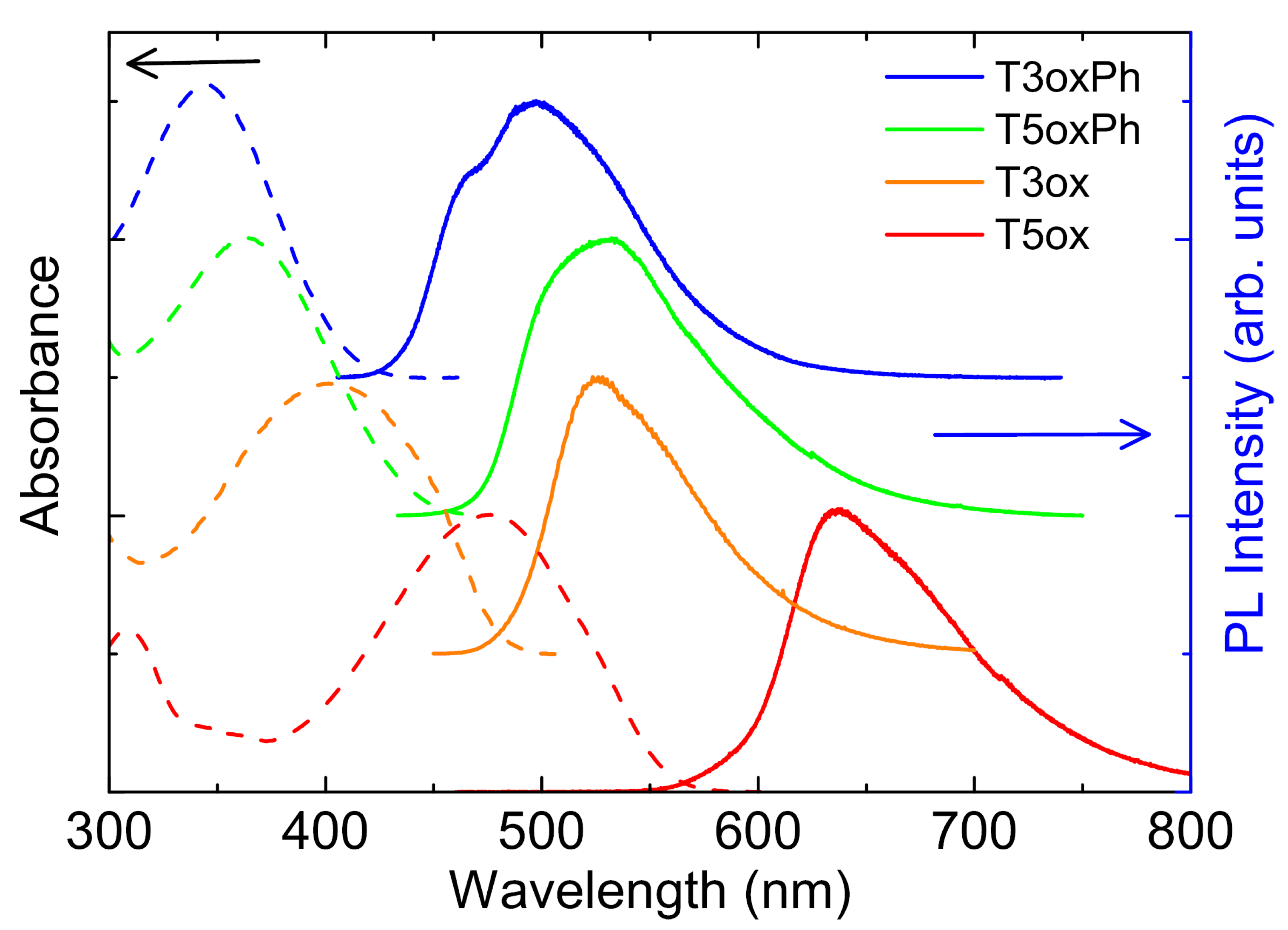
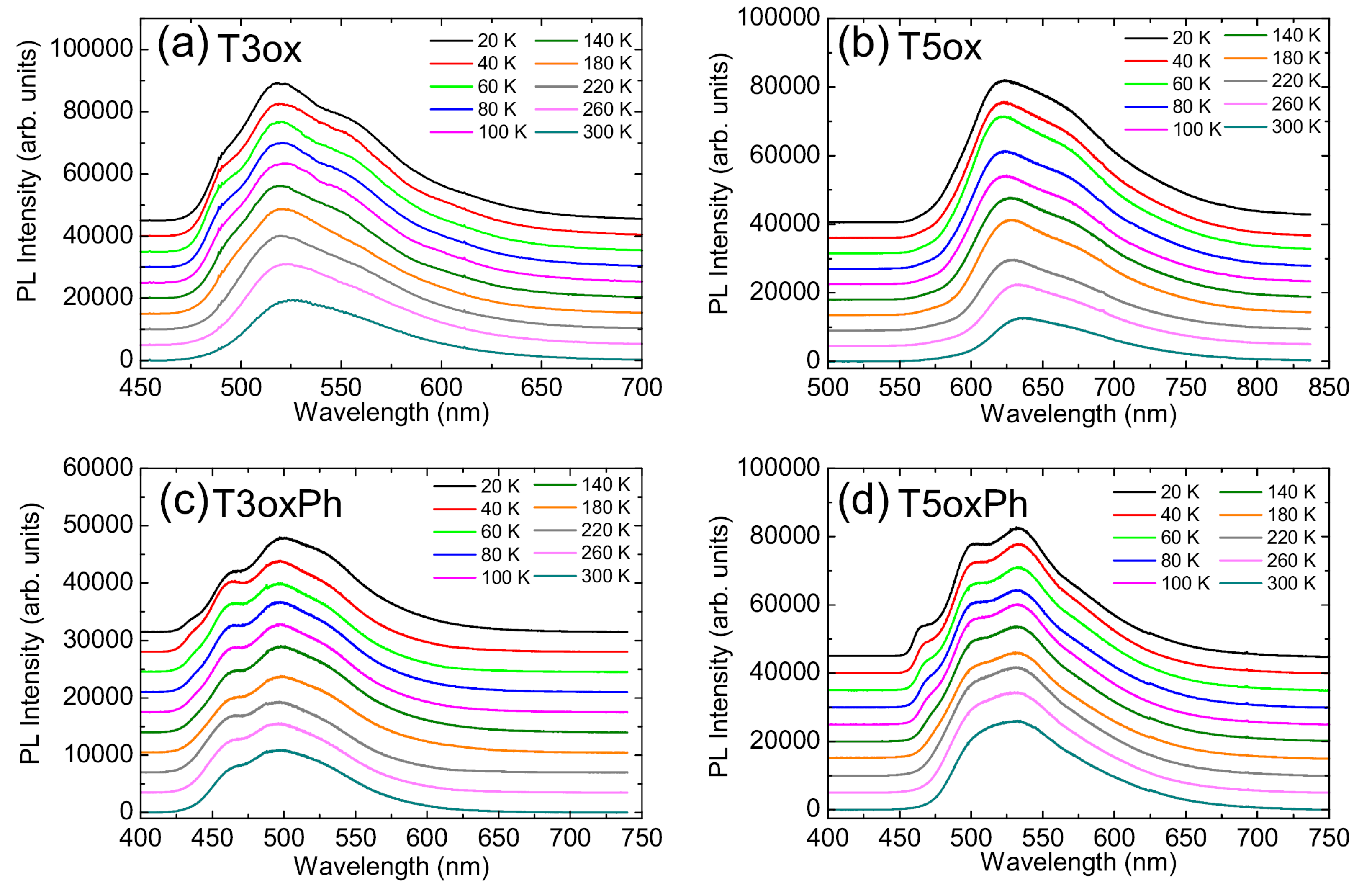
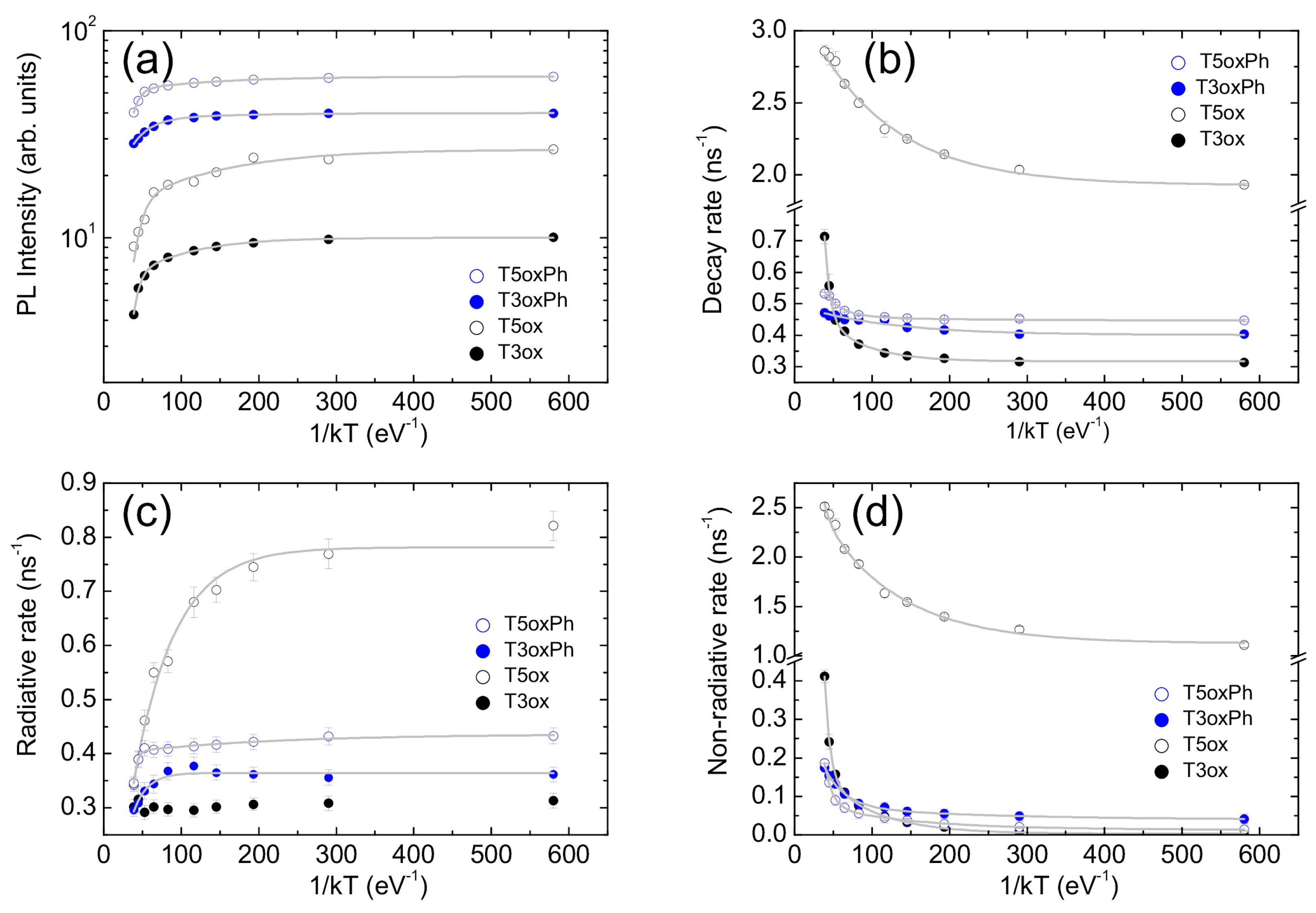
2. Results




3. Conclusions
4. Materials and Methods
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Chen, L.X. Organic Solar Cells: Recent Progress and Challenges. ACS Energy Lett. 2019, 4, 2537–2539. [Google Scholar] [CrossRef] [Green Version]
- Ren, H.; Chen, J.D.; Li, Y.Q.; Tang, J.X. Recent Progress in Organic Photodetectors and their Applications. Adv. Sci. 2021, 8, 2002418. [Google Scholar] [CrossRef]
- Chang, J.; Lin, Z.; Zhang, C.; Hao, Y. Organic Field-Effect Transistor: Device Physics, Materials, and Process. In Different Types of Field-Effect Transistors; Pejovic, M.M., Pejovic, M.M., Eds.; IntechOpen: Rijeka, Croatia, 2017; Chapter 7. [Google Scholar] [CrossRef] [Green Version]
- Zou, S.J.; Shen, Y.; Xie, F.M.; Chen, J.D.; Li, Y.Q.; Tang, J.X. Recent advances in organic light-emitting diodes: Toward smart lighting and displays. Mater. Chem. Front. 2020, 4, 788–820. [Google Scholar] [CrossRef]
- Anni, M.; Lattante, S. Organic Lasers: Fundamentals, Developments, and Applications; Pan Stanford Publishing: Singapore, 2018; pp. 1–324. [Google Scholar]
- Zhang, Q.; Tao, W.; Huang, J.; Xia, R.; Cabanillas-Gonzalez, J. Toward Electrically Pumped Organic Lasers: A Review and Outlook on Material Developments and Resonator Architectures. Adv. Photonics Res. 2021, 2, 2000155. [Google Scholar] [CrossRef]
- Yuvaraja, S.; Nawaz, A.; Liu, Q.; Dubal, D.; Surya, S.G.; Salama, K.N.; Sonar, P. Organic field-effect transistor-based flexible sensors. Chem. Soc. Rev. 2020, 49, 3423–3460. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Liu, Z. Recent progress in organic field-effect transistor-basedbchem/bio-sensors. View 2022, 3, 20200115. [Google Scholar] [CrossRef]
- Gillanders, R.N.; Glackin, J.M.; Filipi, J.; Kezic, N.; Samuel, I.D.; Turnbull, G.A. Preconcentration techniques for trace explosive sensing. Sci. Total Environ. 2019, 658, 650–658. [Google Scholar] [CrossRef] [Green Version]
- Capobianco, M.L.; Barbarella, G.; Manetto, A. Oligothiophenes as Fluorescent Markers for Biological Applications. Molecules 2012, 17, 910–933. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.Q.; Doan, V.; Schwartz, B.J. Conjugated polymer aggregates in solution: Control of interchain interactions. J. Chem. Phys. 1999, 110, 4068–4078. [Google Scholar] [CrossRef]
- Belletete, M.; Bouchard, J.; Leclerc, M.; Durocher, G. Photophysics and Solvent-Induced Aggregation of 2,7-Carbazole-Based Conjugated Polymers. Macromolecules 2005, 38, 880–887. [Google Scholar] [CrossRef]
- Palsson, L.O.; Wang, C.; Russell, D.L.; Monkman, A.P.; Bryce, M.R.; Rumbles, G.; Samuel, I.D. Photophysics of a fluorene co-polymer in solution and films. Chem. Phys. 2002, 279, 229–237. [Google Scholar] [CrossRef]
- Luo, J.; Xie, Z.; Lam, J.W.Y.; Cheng, L.; Chen, H.; Qiu, C.; Kwok, H.S.; Zhan, X.; Liu, Y.; Zhu, D.; et al. Aggregation-induced emission of 1-methyl-1,2,3,4,5-pentaphenylsilole. Chem. Commun. 2001, 1740–1741. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Lam, J.W.Y.; Tang, B.Z. Aggregation-induced emission. Chem. Soc. Rev. 2011, 40, 5361–5388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mei, J.; Leung, N.L.C.; Kwok, R.T.K.; Lam, J.W.Y.; Tang, B.Z. Aggregation-Induced Emission: Together We Shine, United We Soar! Chem. Rev. 2015, 115, 11718–11940. [Google Scholar] [CrossRef] [PubMed]
- Shen, P.; Zhuang, Z.; Zhao, Z.; Tang, B.Z. AIEgens based on main group heterocycles. J. Mater. Chem. C 2018, 6, 11835–11852. [Google Scholar] [CrossRef]
- Granström, M.; Harrison, M.G.; Friend, R.H. Handbook of Oligo- and Polythiophenes; Wiley VHC: Weinheim, Germany, 1999. [Google Scholar]
- Rasmussen, S.C.; Ogawa, K.; Rothstein, S.D. Handbook of Organic Electronics and Photonics; American Scientific Publisher: Valencia, CA, USA, 2008; Chapter 1. [Google Scholar]
- Barbarella, G.; Melucci, M.; Sotgiu, G. The Versatile Thiophene: An Overview of Recent Research on Thiophene-Based Materials. Adv. Mater. 2005, 17, 1581–1593. [Google Scholar] [CrossRef]
- Turkoglu, G.; Cinar, M.E.; Ozturk, T. Thiophene-Based Organic Semiconductors. Top. Curr. Chem. 2017, 375, 84. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.C.; Evenson, S.J.; McCausland, C.B. Fluorescent thiophene-based materials and their outlook for emissive applications. Chem. Commun. 2015, 51, 4528–4543. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Kan, B.; Liu, F.; Long, G.; Wan, X.; Chen, X.; Zuo, Y.; Ni, W.; Zhang, H.; Li, M.; et al. Small-molecule solar cells with efficiency over 9%. Nat. Photonics 2015, 9, 35–41. [Google Scholar] [CrossRef]
- Kan, B.; Li, M.; Zhang, Q.; Liu, F.; Wan, X.; Wang, Y.; Ni, W.; Long, G.; Yang, X.; Feng, H.; et al. A Series of Simple Oligomer-like Small Molecules Based on Oligothiophenes for Solution-Processed Solar Cells with High Efficiency. J. Am. Chem. Soc. 2015, 137, 3886–3893. [Google Scholar] [CrossRef]
- Wu, J.; Li, G.; Fang, J.; Guo, X.; Zhu, L.; Guo, B.; Wang, Y.; Zhang, G.; Arunagiri, L.; Liu, F.; et al. Random terpolymer based on thiophene-thiazolothiazole unit enabling efficient non-fullerene organic solar cells. Nat. Commun. 2020, 11, 4612. [Google Scholar] [CrossRef]
- Horowitz, G.; Hajlaoui, M.E. Mobility in Polycrystalline Oligothiophene Field-Effect Transistors Dependent on Grain Size. Adv. Mater. 2000, 12, 1046–1050. [Google Scholar] [CrossRef]
- Amna, B.; Isci, R.; Siddiqi, H.M.; Majewski, L.A.; Faraji, S.; Ozturk, T. High-performance, low-voltage organic field-effect transistors using thieno[3,2-b]thiophene and benzothiadiazole co-polymers. J. Mater. Chem. C 2022, 10, 8254–8265. [Google Scholar] [CrossRef]
- Larik, F.A.; Faisal, M.; Saeed, A.; Abbas, Q.; Kazi, M.A.; Abbas, N.; Thebo, A.A.; Khan, D.M.; Channar, P.A. Thiophene-based molecular and polymeric semiconductors for organic field effect transistors and organic thin film transistors. J. Mater. Sci. Mater. Electron. 2018, 29, 17975–18010. [Google Scholar] [CrossRef]
- Kanemitsu, Y.; Suzuki, K.; Masumoto, Y.; Tomiuchi, Y.; Shiraishi, Y.; Kuroda, M. Optical properties of quasi-one-dimensional thiophene-based oligomers. Phys. Rev. B 1994, 50, 2301–2305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yassar, A.; Horowitz, G.; Valat, P.; Wintgens, V.; Hmyene, M.; Deloffre, F.; Srivastava, P.; Lang, P.; Garnier, F. Exciton Coupling Effects in the Absorption and Photoluminescence of Sexithiophene Derivatives. J. Phys. Chem. 1995, 99, 9155–9159. [Google Scholar] [CrossRef]
- Perepichka, I.; Perepichka, D.F.; Meng, H.; Wudl, F. Light-Emitting Polythiophenes. Adv. Mater. 2005, 17, 2281–2305. [Google Scholar] [CrossRef]
- Grimsdale, A.C.; Leok Chan, K.; Martin, R.E.; Jokisz, P.G.; Holmes, A.B. Synthesis of Light-Emitting Conjugated Polymers for Applications in Electroluminescent Devices. Chem. Rev. 2009, 109, 897–1091. [Google Scholar] [CrossRef]
- Becker, R.S.; Seixas de Melo, J.; Macanita, A.L.; Elisei, F. Comprehensive Evaluation of the Absorption, Photophysical, Energy Transfer, Structural, and Theoretical Properties of α-Oligothiophenes with One to Seven Rings. J. Phys. Chem. 1996, 100, 18683–18695. [Google Scholar] [CrossRef] [Green Version]
- Beljonne, D.; Shuai, Z.; Pourtois, G.; Bredas, J.L. Spin-Orbit Coupling and Intersystem Crossing in Conjugated Polymers: A Configuration Interaction Description. J. Phys. Chem. A 2001, 105, 3899–3907. [Google Scholar] [CrossRef]
- Barbarella, G.; Favaretto, L.; Sotgiu, G.; Zambianchi, M.; Bongini, A.; Arbizzani, C.; Mastragostino, M.; Anni, M.; Gigli, G.; Cingolani, R. Tuning Solid-State Photoluminescence Frequencies and Efficiencies of Oligomers Containing One Central Thiophene-S,S-dioxide Unit. J. Am. Chem. Soc. 2000, 122, 11971–11978. [Google Scholar] [CrossRef]
- Tsai, C.H.; Chirdon, D.N.; Maurer, A.B.; Bernhard, S.; Noonan, K.J.T. Synthesis of Thiophene 1,1-Dioxides and Tuning Their Optoelectronic Properties. Org. Lett. 2013, 15, 5230–5233. [Google Scholar] [CrossRef]
- Barbarella, G.; Favaretto, L.; Sotgiu, G.; Zambianchi, M.; Fattori, V.; Cocchi, M.; Cacialli, F.; Gigli, G.; Cingolani, R. Modified Oligothiophenes with High Photo- and Electroluminescence Efficiencies. Adv. Mater. 1999, 11, 1375–1379. [Google Scholar] [CrossRef]
- Anni, M.; Gigli, G.; Paladini, V.; Cingolani, R.; Barbarella, G.; Favaretto, L.; Sotgiu, G.; Zambianchi, M. Color engineering by modified oligothiophene blends. Appl. Phys. Lett. 2000, 77, 2458–2460. [Google Scholar] [CrossRef]
- Barbarella, G.; Favaretto, L.; Zambianchi, M.; Pudova, O.; Arbizzani, C.; Bongini, A.; Mastragostino, M. From Easily Oxidized to Easily Reduced Thiophene-Based Materials. Adv. Mater. 1998, 10, 551–554. [Google Scholar] [CrossRef]
- Barbarella, G.; Favaretto, L.; Sotgiu, G.; Antolini, L.; Gigli, G.; Cingolani, R.; Bongini, A. Rigid-Core Oligothiophene-S,S-dioxides with High Photoluminescence Efficiencies Both in Solution and in the Solid State. Chem. Mater. 2001, 13, 4112–4122. [Google Scholar] [CrossRef]
- Anni, M.; Della Sala, F.; Raganato, M.F.; Fabiano, E.; Lattante, S.; Cingolani, R.; Gigli, G.; Barbarella, G.; Favaretto, L.; Görling, A. Nonradiative Relaxation in Thiophene-S,S-dioxide Derivatives: The Role of the Environment. J. Phys. Chem. B 2005, 109, 6004–6011. [Google Scholar] [CrossRef]
- Xie, L.H.; Hou, X.Y.; Hua, Y.R.; Huang, Y.Q.; Zhao, B.M.; Liu, F.; Peng, B.; Wei, W.; Huang, W. An Effective Strategy to Tune Supramolecular Interaction via a Spiro-Bridged Spacer in Oligothiophene-S,S-dioxides and Their Anomalous Photoluminescent Behavior. Org. Lett. 2007, 9, 1619–1622. [Google Scholar] [CrossRef]
- Osken, I.; Gundogan, A.S.; Tekin, E.; Eroglu, M.S.; Ozturk, T. Fluorene-Dithienothiophene-S,S-dioxide Copolymers. Fine-Tuning for OLED Applications. Macromolecules 2013, 46, 9202–9210. [Google Scholar] [CrossRef]
- Barbarella, G.; Zambianchi, M.; Antolini, L.; Ostoja, P.; Maccagnani, P.; Bongini, A.; Marseglia, E.A.; Tedesco, E.; Gigli, G.; Cingolani, R. Solid-State Conformation, Molecular Packing, and Electrical and Optical Properties of Processable β-Methylated Sexithiophenes. J. Am. Chem. Soc. 1999, 121, 8920–8926. [Google Scholar] [CrossRef]
- Antolini, L.; Tedesco, E.; Barbarella, G.; Favaretto, L.; Sotgiu, G.; Zambianchi, M.; Casarini, D.; Gigli, G.; Cingolani, R. Molecular Packing and Photoluminescence Efficiency in Odd-Membered Oligothiophene S,S-Dioxides. J. Am. Chem. Soc. 2000, 122, 9006–9013. [Google Scholar] [CrossRef]
- Lanzani, G.; Cerullo, G.; De Silvestri, S.; Barbarella, G.; Sotgiu, G. Influence of the environment on the excited state deactivation in functionalized quinque-thienyls. J. Chem. Phys. 2001, 115, 1623–1625. [Google Scholar] [CrossRef]
- Lattante, S.; De Giorgi, M.; Barbarella, G.; Favaretto, L.; Gigli, G.; Cingolani, R.; Anni, M. Interplay between stimulated emission and singlet-singlet annihilation in oligothiophene dioxide thin films. J. Appl. Phys. 2006, 100, 023530. [Google Scholar] [CrossRef]
- Anni, M.; Lattante, S.; Cingolani, R.; Gigli, G.; Barbarella, G.; Favaretto, L. Far-field emission and feedback origin of random lasing in oligothiophene dioxide neat films. Appl. Phys. Lett. 2003, 83, 2754–2756. [Google Scholar] [CrossRef]
- Anni, M.; Lattante, S.; Stomeo, T.; Cingolani, R.; Gigli, G.; Barbarella, G.; Favaretto, L. Modes interaction and light transport in bidimensional organic random lasers in the weak scattering limit. Phys. Rev. B 2004, 70, 195216. [Google Scholar] [CrossRef]
- Ghofraniha, N.; Viola, I.; Di Maria, F.; Barbarella, G.; Gigli, G.; Leuzzi, L.; Conti, C. Experimental evidence of replica symmetry breaking in random lasers. Nat. Commun. 2015, 6, 6058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, J.; Li, S.; Wang, H.C.; Bai, S.J.; Wang, Z.; Ren, X.; Xu, Y.X. Distinct luminescent properties between thiophene-S-oxide and Thiophene-S, S-dioxides incorporated ladder-type molecules. Dyes Pigm. 2020, 175, 108147. [Google Scholar] [CrossRef]
- Nakahama, T.; Kitagawa, D.; Sotome, H.; Ito, S.; Miyasaka, H.; Kobatake, S. Optical properties and solvatofluorochromism of fluorene derivatives bearing S,S-dioxidized thiophene. Photochem. Photobiol. Sci. 2016, 15, 1254–1263. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhang, H.; Luo, W.; Nie, H.; Hu, R.; Qin, A.; Zhao, Z.; Tang, B.Z. Oxidation-enhanced emission: Exploring novel AIEgens from thieno[3,2-b]thiophene S,S-dioxide. J. Mater. Chem. C 2017, 5, 960–968. [Google Scholar] [CrossRef]
- Sala, F.D.; Heinze, H.; Görling, A. Excitation energies of terthiophene and its dioxide derivative: A first-principles study. Chem. Phys. Lett. 2001, 339, 343–350. [Google Scholar] [CrossRef]
- Della Sala, F.; Gigli, G.; Raganato, M.; Anni, M.; Pisignano, D.; Cingolani, R.; Favaretto, L.; Sotgiu, G.; Barbarella, G.; Antolini, L. Effects of intermolecular interactions on photoluminescence efficiency of crystalline thienylene-S,S-dioxide molecular semiconductors. Org. Electron. 2004, 5, 129–134. [Google Scholar] [CrossRef]
- Gigli, G.; Barbarella, G.; Favaretto, L.; Cacialli, F.; Cingolani, R. High-efficiency oligothiopene-based light-emitting diodes. Appl. Phys. Lett. 1999, 75, 439–441. [Google Scholar] [CrossRef]
- Louarn, G.; Buisson, J.P.; Lefrant, S.; Fichou, D. Vibrational Studies of a Series of .alpha.-Oligothiophenes as Model Systems of Polythiophene. J. Phys. Chem. 1995, 99, 11399–11404. [Google Scholar] [CrossRef]
- Ariu, M.; Sims, M.; Rahn, M.D.; Hill, J.; Fox, A.M.; Lidzey, D.G.; Oda, M.; Cabanillas-Gonzalez, J.; Bradley, D.D.C. Exciton migration in β-phase poly(9,9-dioctylfluorene). Phys. Rev. B 2003, 67, 195333. [Google Scholar] [CrossRef]
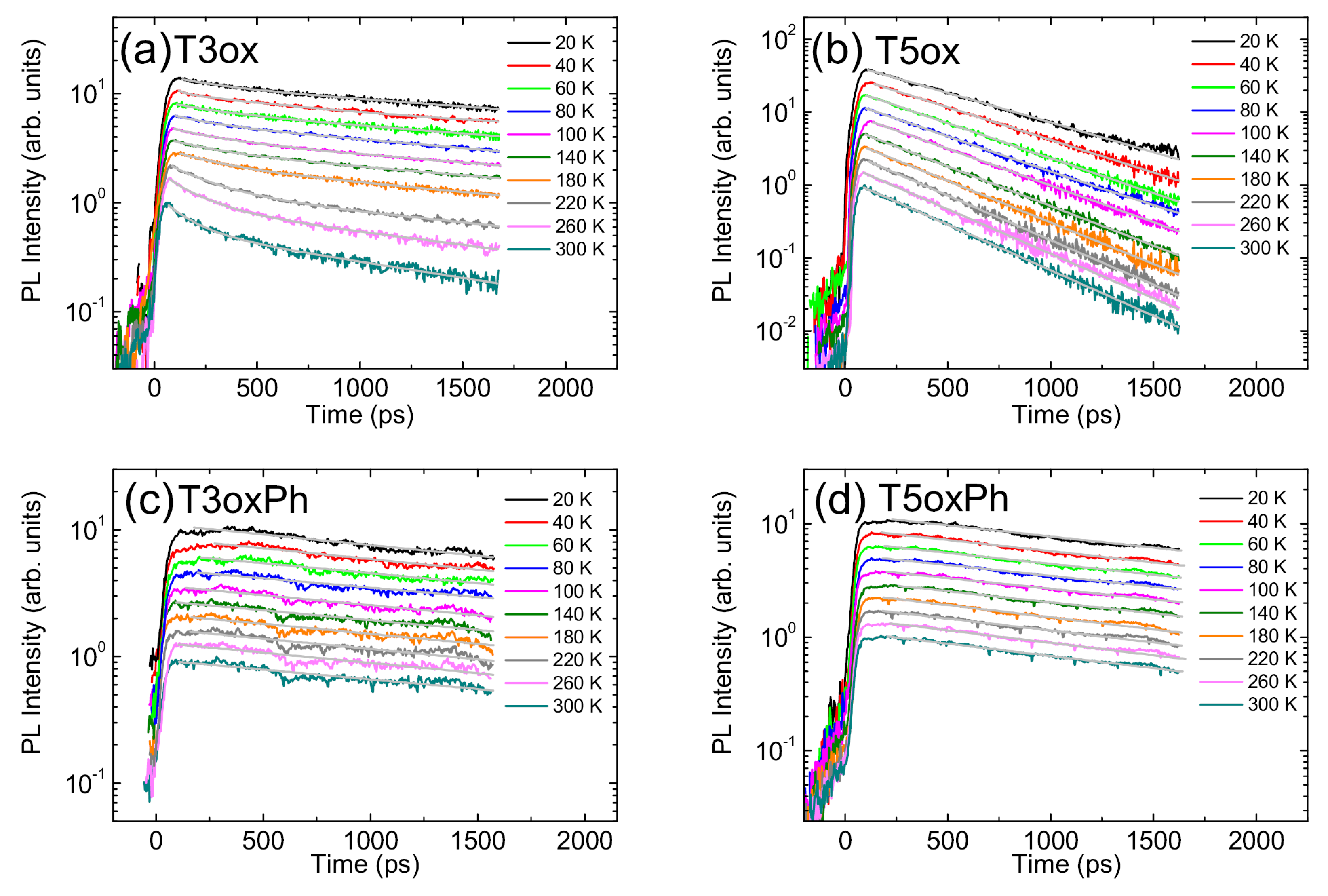
- In this case the temperature dependence of the slow process, related to exciton recombination, has been considered for the following analysis.
- Hermet, P.; Bantignies, J.L.; Maurin, D.; Sauvajol, J.L. Terahertz spectroscopy of the crystalline α-quaterthiophene: A combined experimental and density functional theory study. Chem. Phys. Lett. 2007, 445, 47–50. [Google Scholar] [CrossRef]
- Cirmi, G.; Brida, D.; Gambetta, A.; Piacenza, M.; Sala, F.D.; Favaretto, L.; Cerullo, G.; Lanzani, G. Observation and control of coherent torsional dynamics in a quinquethiophene molecule. Phys. Chem. Chem. Phys. 2010, 12, 7917–7923. [Google Scholar] [CrossRef]
- Johnston, M.; Herz, L.; Khan, A.; Köhler, A.; Davies, A.; Linfield, E. Low-energy vibrational modes in phenylene oligomers studied by THz time-domain spectroscopy. Chem. Phys. Lett. 2003, 377, 256–262. [Google Scholar] [CrossRef]
- Macchi, G.; Medina, B.M.; Zambianchi, M.; Tubino, R.; Cornil, J.; Barbarella, G.; Gierschner, J.; Meinardi, F. Spectroscopic signatures for planar equilibrium geometries in methyl-substituted oligothiophenes. Phys. Chem. Chem. Phys. 2009, 11, 984–990. [Google Scholar] [CrossRef]
- Chenouf, J.; Boutahir, M.; Fakrach, B.; Rahmani, A.; Chadli, H.; Hermet, P.; Mejia-Lopez, J.; Rahmani, A. Encapsulation effect of π-conjugated quaterthiophene on the radial breathing and tangential modes of semiconducting and metallic single-walled carbon nanotubes. J. Comput. Chem. 2020, 41, 2420–2428. [Google Scholar] [CrossRef]
- Moreno Castro, C.; Ruiz Delgado, M.C.; Hernandez, V.; Hotta, S.; Casado, J.; Lopez Navarrete, J.T. Efficiency of the π conjugation in a novel family of α,α’-bisphenyl end-capped oligothiophenes by means of Raman spectroscopy. J. Chem. Phys. 2002, 116, 10419–10427. [Google Scholar] [CrossRef]
- Sharafy, S.; Muszkat, K.A. Viscosity dependence of fluorescence quantum yields. J. Am. Chem. Soc. 1971, 93, 4119–4125. [Google Scholar] [CrossRef]
- Bongini, A.; Barbarella, G.; Favaretto, L.; Sotgiu, G.; Zambianchi, M.; Casarini, D. Conformational profile, energy barriers and optical properties of quinquethiophene-S,S-dioxides. Tetrahedron 2002, 58, 10151–10158. [Google Scholar] [CrossRef]
- Englman, R.; Jortner, J. The energy gap law for radiationless transitions in large molecules. Mol. Phys. 1970, 18, 145–164. [Google Scholar] [CrossRef]
- Greenham, N.; Samuel, I.; Hayes, G.; Phillips, R.; Kessener, Y.; Moratti, S.; Holmes, A.; Friend, R. Measurement of absolute photoluminescence quantum efficiencies in conjugated polymers. Chem. Phys. Lett. 1995, 241, 89–96. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Decay Time (ns) | PLQY (%) | Radiative Time (ns) | Non-Radiative Time (ns) |
|---|---|---|---|---|
| T3ox | 1.65 ± 0.05 | 46.0 ± 1.4 | 3.60 ± 0.18 | 3.10 ± 0.18 |
| T3oxPh | 2.13 ± 0.05 | 63 ± 2 | 3.38 ± 0.14 | 5.8 ± 0.5 |
| T5ox | 0.350 ± 0.010 | 12.0 ± 0.4 | 2.92 ± 0.13 | 0.398 ± 0.013 |
| T5oxPh | 1.88 ± 0.02 | 70 ± 2 | 2.90 ± 0.11 | 5.4 ± 0.3 |
| Molecule | E (meV) | E (meV) | E (meV) | E (meV) | E (meV) | E (meV) | E (meV) | E (meV) |
|---|---|---|---|---|---|---|---|---|
| T3ox | 14.7 ± 1.6 | 148 ± 21 | 19 ± 4 | 150 ± 30 | - | - | 15.0 ± 1.3 | 145 ± 11 |
| T3oxPh | 9 ± 3 | 48 ± 6 | 8.9 ± 1.6 | - | 55 ± 13 | - | 10 ± 2 | 39 ± 6 |
| T5ox | 7 ± 2 | 45 ± 10 | 9.5 ± 0.3 | - | 6 ± 2 | 36 ± 5 | 8 ± 2 | 34 ± 6 |
| T5oxPh | 8.5 ± 1.2 | 50 ± 6 | 39 ± 5 | - | 1.5 ± 0.4 | 35 ± 4 | 8.2 ± 1.1 | 39 ± 6 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anni, M. Investigation of the Origin of High Photoluminescence Quantum Yield in Thienyl-S,S-dioxide AIEgens Oligomers by Temperature Dependent Optical Spectroscopy. Molecules 2023, 28, 5161. https://doi.org/10.3390/molecules28135161
Anni M. Investigation of the Origin of High Photoluminescence Quantum Yield in Thienyl-S,S-dioxide AIEgens Oligomers by Temperature Dependent Optical Spectroscopy. Molecules. 2023; 28(13):5161. https://doi.org/10.3390/molecules28135161
Chicago/Turabian StyleAnni, Marco. 2023. "Investigation of the Origin of High Photoluminescence Quantum Yield in Thienyl-S,S-dioxide AIEgens Oligomers by Temperature Dependent Optical Spectroscopy" Molecules 28, no. 13: 5161. https://doi.org/10.3390/molecules28135161
APA StyleAnni, M. (2023). Investigation of the Origin of High Photoluminescence Quantum Yield in Thienyl-S,S-dioxide AIEgens Oligomers by Temperature Dependent Optical Spectroscopy. Molecules, 28(13), 5161. https://doi.org/10.3390/molecules28135161