
Electrochemical Iodination through the In Situ Generation of Iodinating Agents: A Promising Green Approach

 , ,
, ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
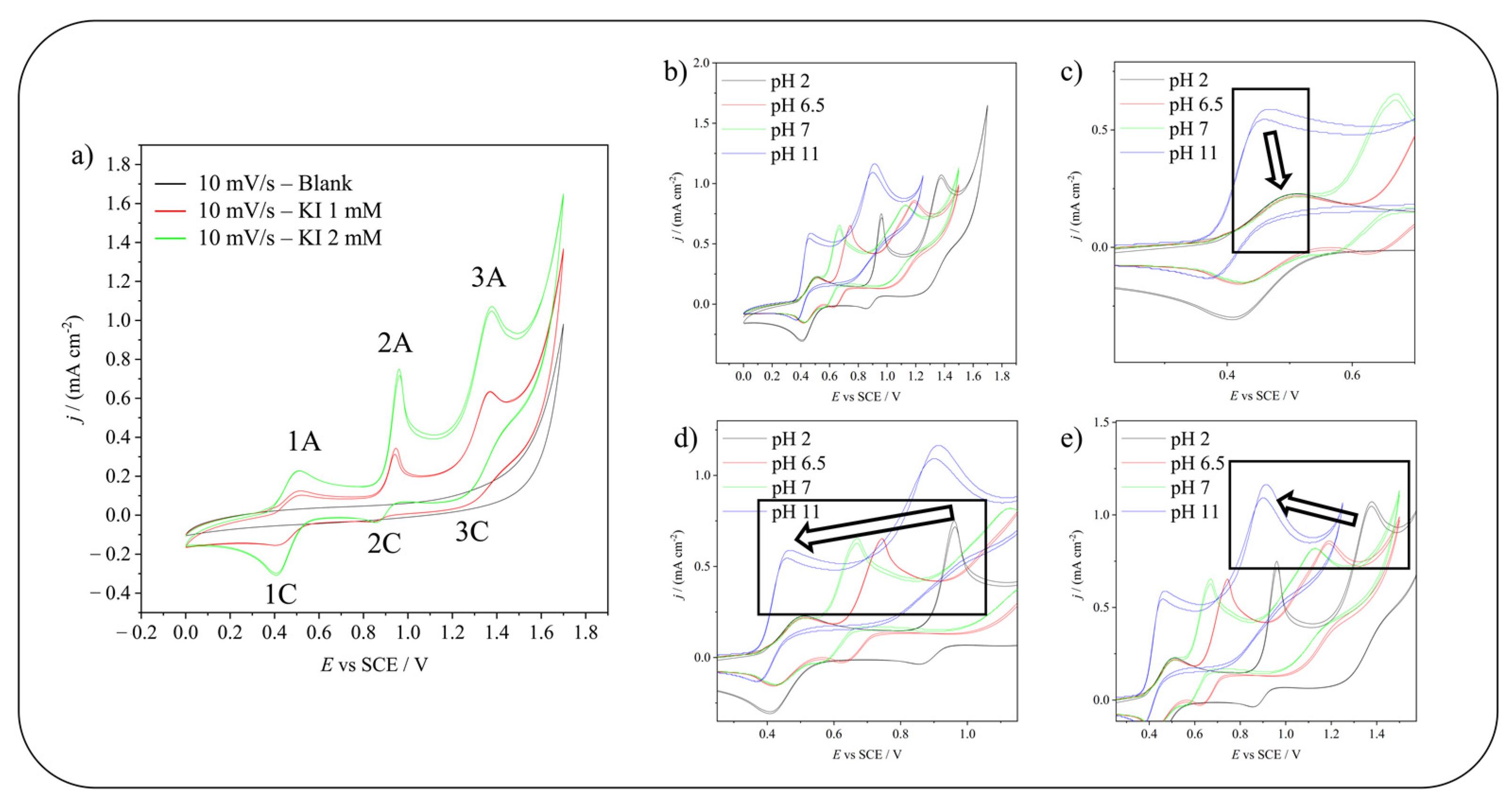
2.1. Electrochemical Oxidation of Iodide/Iodine in Aqueous Solvent on Carbonaceous Materials
| 1A | 2 I− → I2 + 2 e− | Evs SHE (V) = 0.621 | (3) |
| 2 I3−→ 3 I2 + 2 e− | Evs SHE (V) = 0.789 | (4) | |
| 3 I− → I3− + 2 e− | Evs SHE (V) = 0.536 | (5) | |
| 2 I− → I2 + 2 e− | Evs SHE (V) = 0.621 | (6) | |
| 2A | I2 + 2 H2O → 2 HIO + 2 H+ + 2 e− | Evs SHE (V) = 1.354−0.0591 pH | (7) |
| 3A | I− + 3 H2O → HIO3 + 5 H+ + 6 e− | Evs SHE (V) = 1.077−0.0493 pH | (8) |
2.2. Electrochemical Iodination of Phenolic Substrates in Aqueous Solvent on Carbonaceous Materials—CVs Monitoring
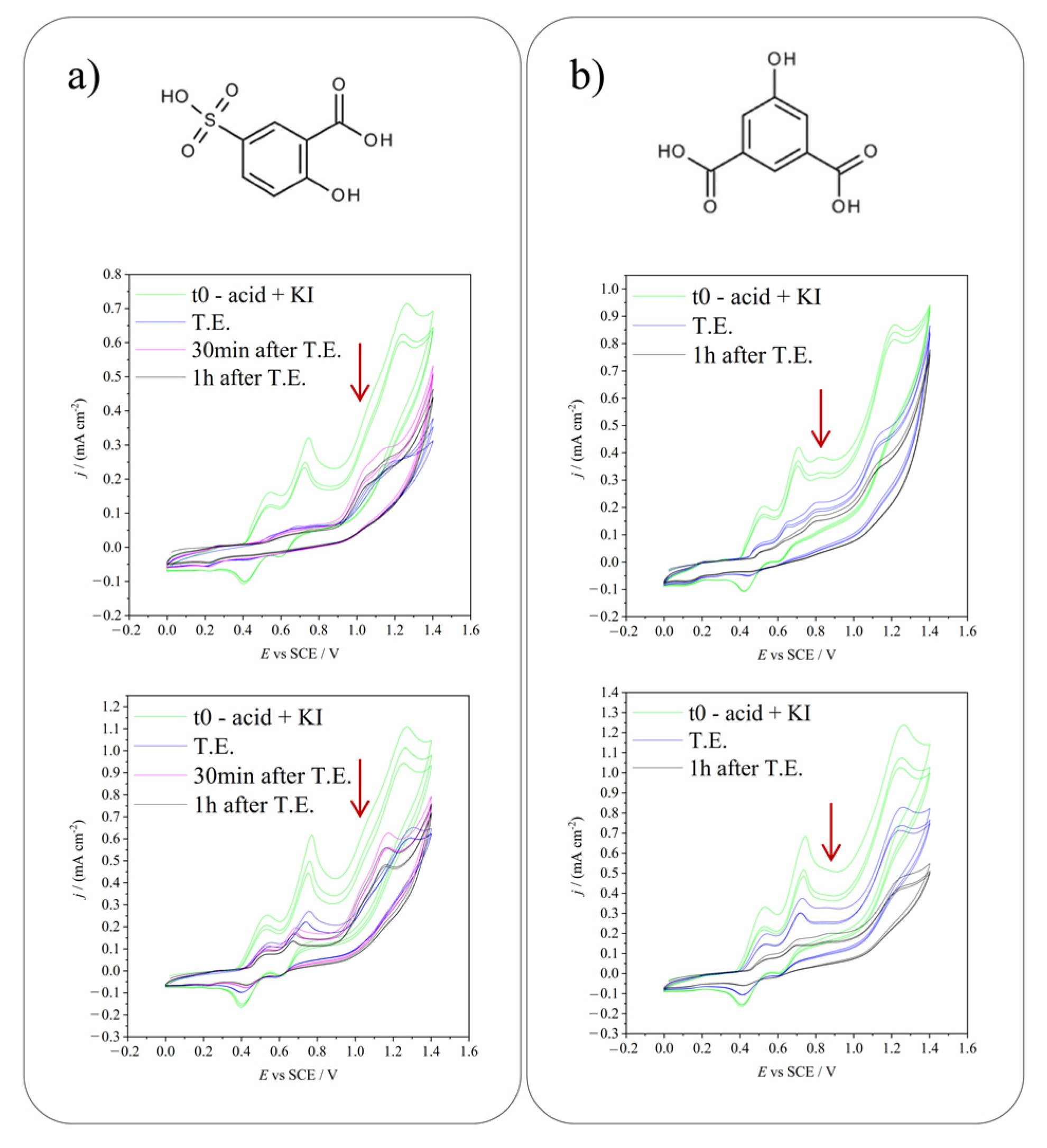
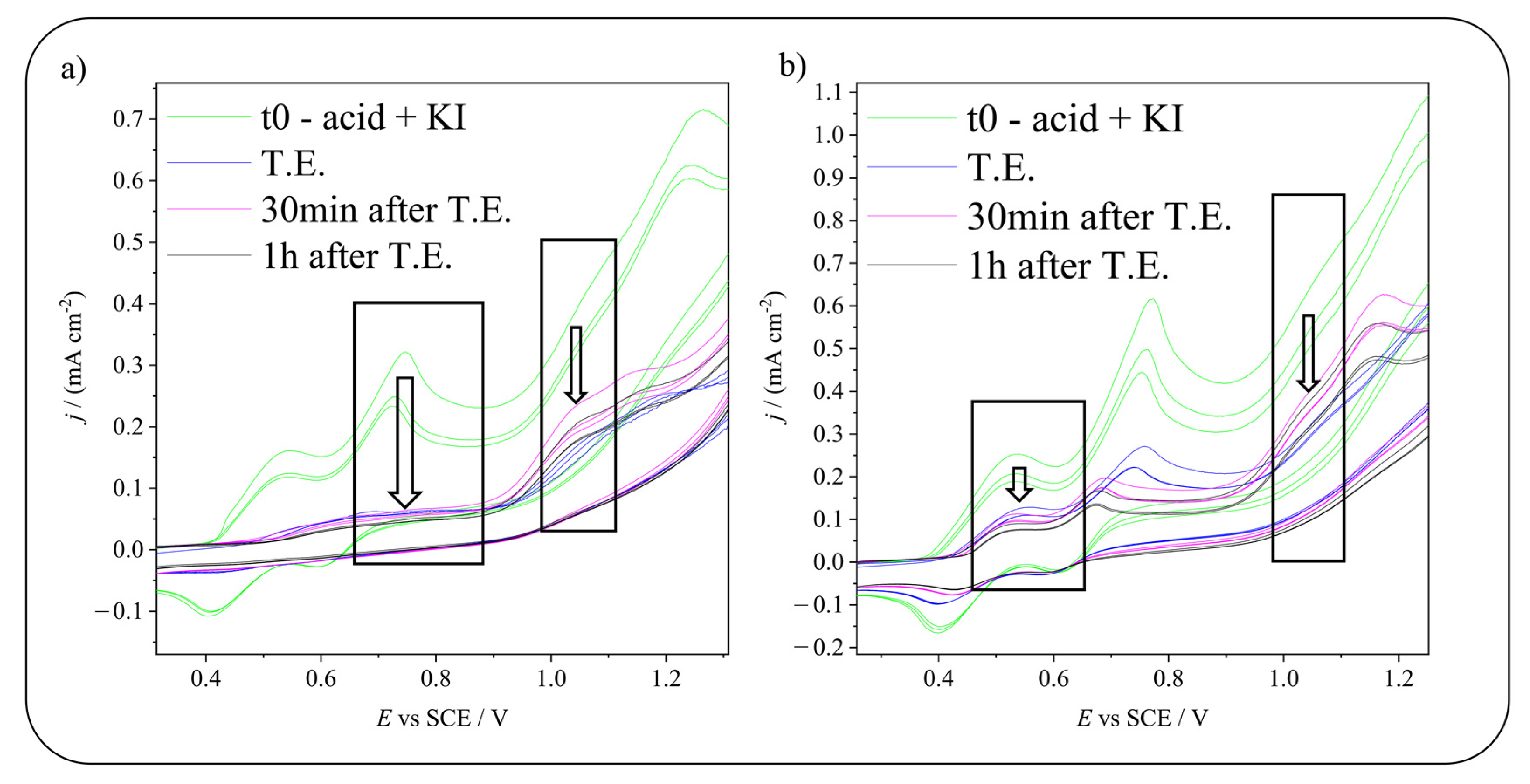
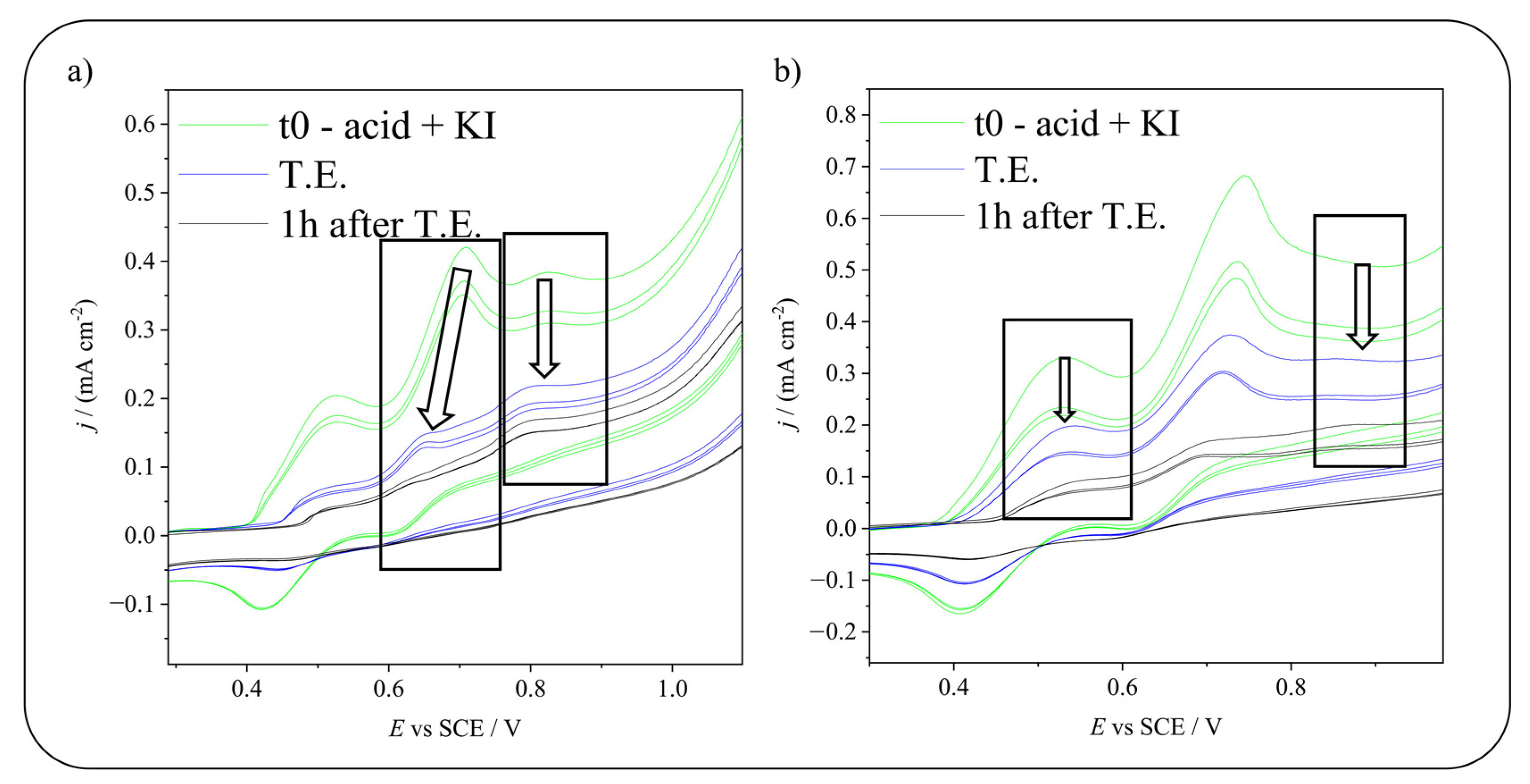
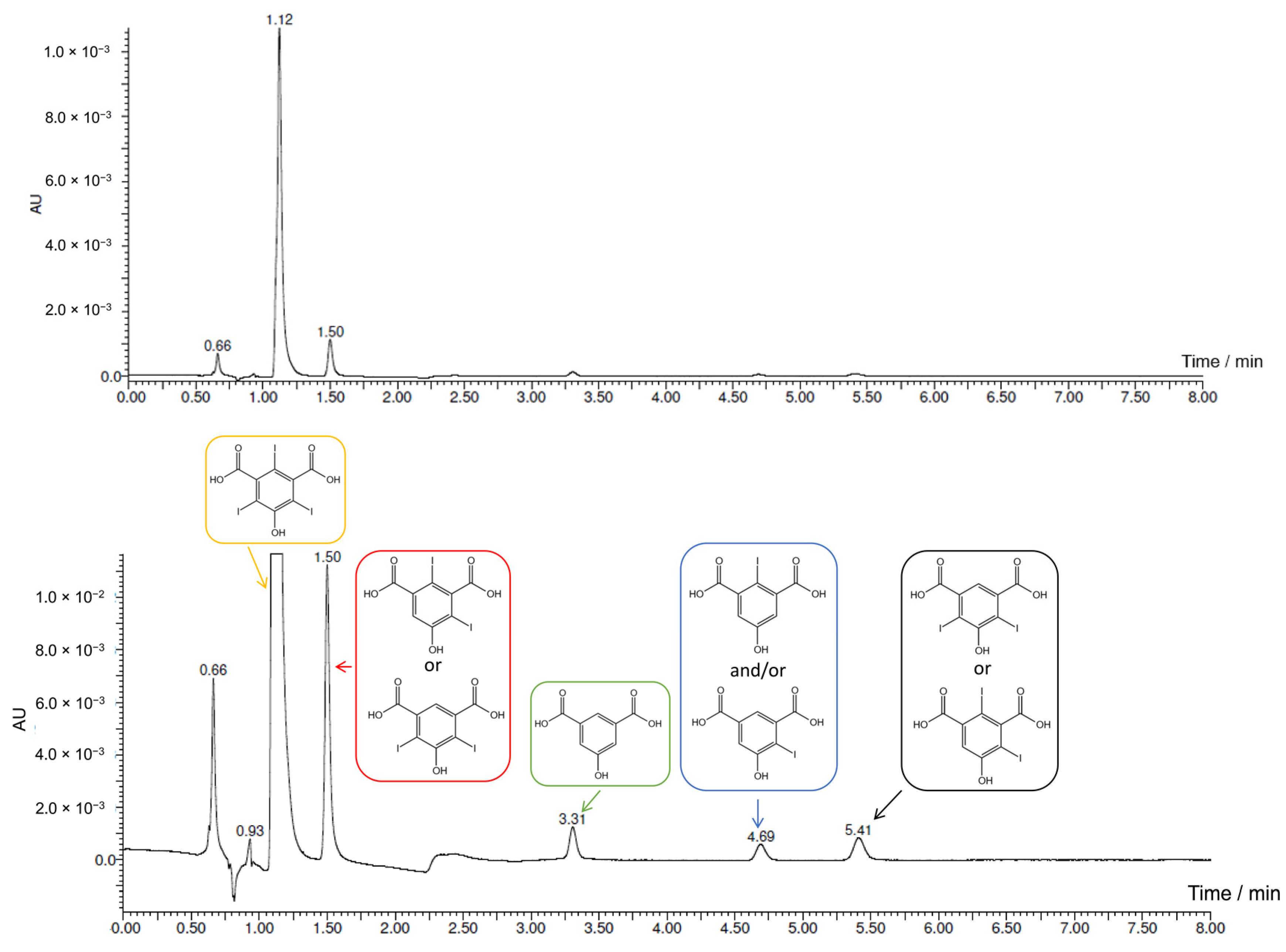
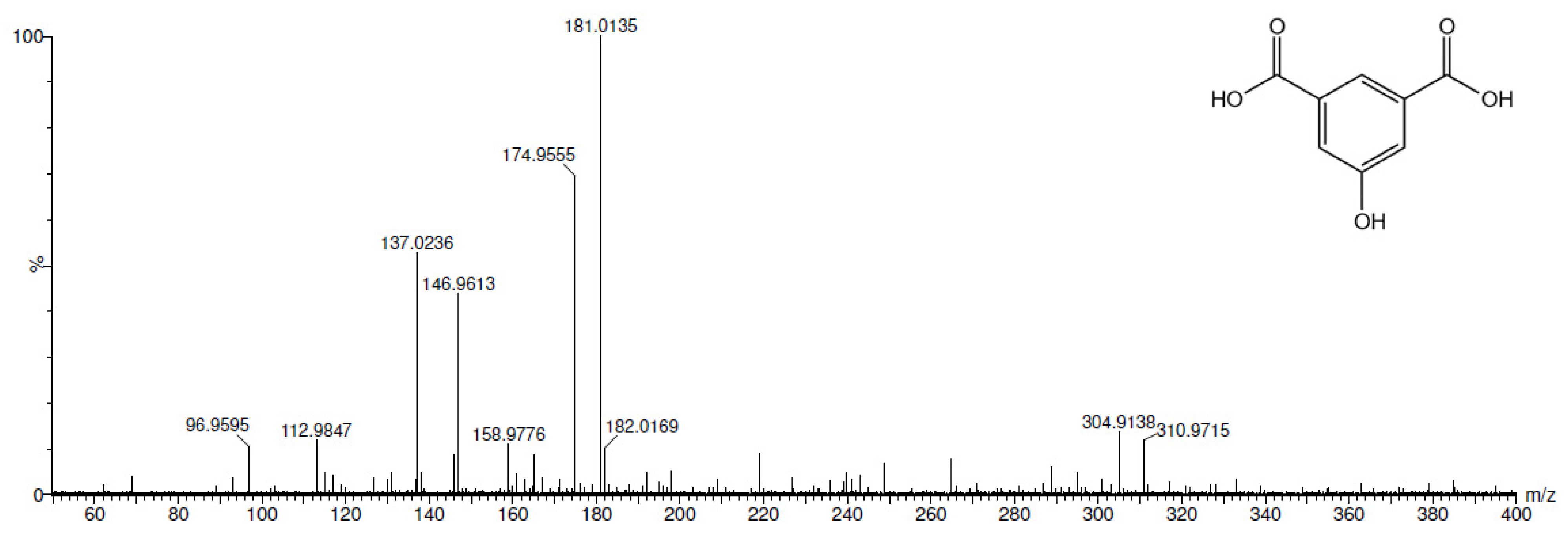
2.3. Electrochemical Iodination of 5-Hydroxyisophthalic Acid—Product Analysis
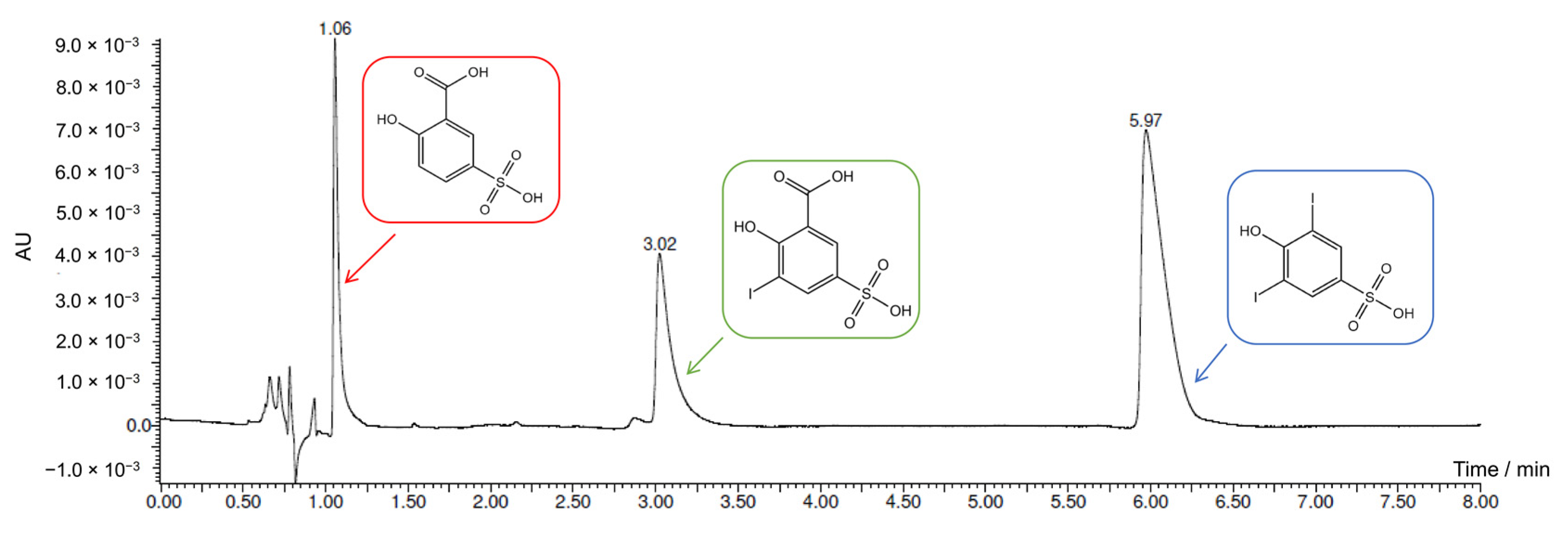
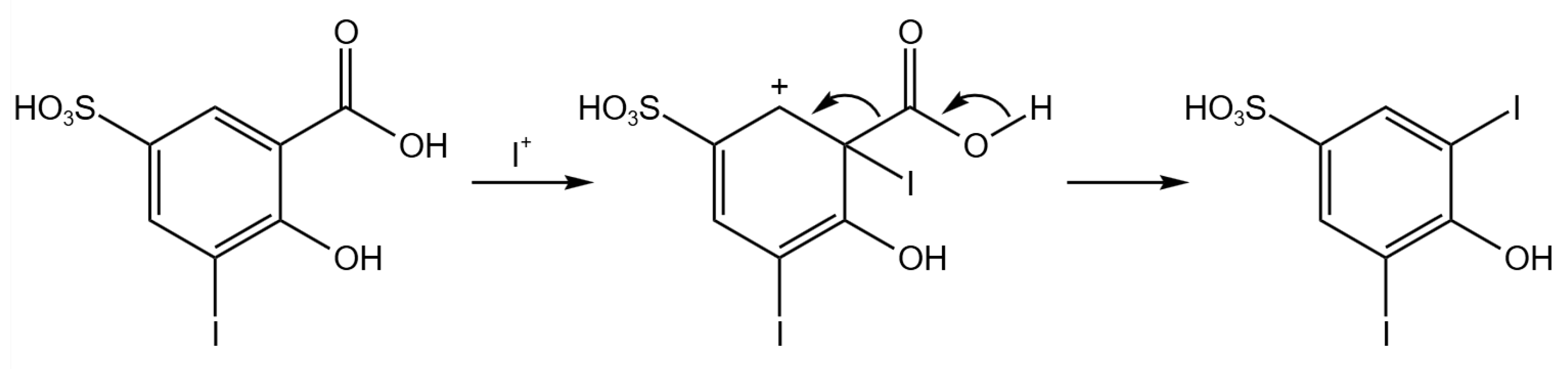
2.4. Electrochemical Iodination of 5-Sulfosalicylic Acid—Product Analysis
3. Materials and Methods
3.1. Chemicals
3.2. Electrochemistry
3.3. UPLC-PDA-HR-MS and NMR
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Drogui, P.; Blais, J.-F.; Mercier, G. Review of Electrochemical Technologies for Environmental Applications. Recent Pat. Eng. 2008, 1, 257–272. [Google Scholar] [CrossRef]
- Chaplin, B.P. The Prospect of Electrochemical Technologies Advancing Worldwide Water Treatment. Acc. Chem. Res. 2019, 52, 596–604. [Google Scholar] [CrossRef]
- Frontana-Uribe, B.A.; Little, R.D.; Ibanez, J.G.; Palma, A.; Vasquez-Medrano, R. Organic electrosynthesis: A promising green methodology in organic chemistry. Green Chem. 2010, 12, 2099–2119. [Google Scholar] [CrossRef]
- Fors, B.P.; Davis, N.R.; Buchwald, S.L. An efficient process for Pd-catalyzed C-N cross-coupling reactions of aryl iodides: Insight into controlling factors. J. Am. Chem. Soc. 2009, 131, 5766–5768. [Google Scholar] [CrossRef] [Green Version]
- Kinbara, A.; Ito, M.; Abe, T.; Yamagishi, T. Nickel-catalyzed C-P cross-coupling reactions of aryl iodides with H-phosphinates. Tetrahedron 2015, 71, 7614–7619. [Google Scholar] [CrossRef]
- Bonaterra, M.; Martín, S.E.; Rossi, R.A. One-pot palladium-catalyzed cross-coupling reaction of aryl iodides with stannylarsanes and stannylstibanes. Org. Lett. 2003, 5, 2731–2734. [Google Scholar] [CrossRef]
- Day, D.P.; Alsenani, N.I.; Alsimaree, A.A. Reactivity and Applications of Iodine Monochloride in Synthetic Approaches. Eur. J. Org. Chem. 2021, 2021, 4299–4307. [Google Scholar] [CrossRef]
- Bergström, M.; Suresh, G.; Naidu, V.R.; Unelius, C.R. N-Iodosuccinimide (NIS) in Direct Aromatic Iodination. Eur. J. Org. Chem. 2017, 2017, 3234–3239. [Google Scholar] [CrossRef]
- Taguchi, H. Iodinating Reagents. In Iodine Chemistry and Applications; Wiley Online Library: Hoboken, NJ, USA, 2015; pp. 249–276. [Google Scholar] [CrossRef]
- Chaikovskii, V.K.; Filimonov, V.D.; Funk, A.A.; Skorokhodov, V.I.; Ogorodnikov, V.D. 1,3-Diiodo-5,5-dimethylhydantoin-An efficient reagent for iodination of aromatic compounds. Russ. J. Org. Chem. 2007, 43, 1291–1296. [Google Scholar] [CrossRef]
- Reddy, K.S.K.; Narender, N.; Rohitha, C.N.; Kulkarni, S.J. Iodination of aromatic compounds using potassium iodide and hydrogen peroxide. Synth. Commun. 2008, 38, 3894–3902. [Google Scholar] [CrossRef]
- Scheide, M.R.; Nicoleti, C.R.; Martins, G.M.; Braga, A.L. Electrohalogenation of organic compounds. Org. Biomol. Chem. 2021, 19, 2578–2602. [Google Scholar] [CrossRef]
- Miller, L.L.; Kujawa, E.P.; Campbell, C.B. Iodination with Electrolytically Generated Iodine(I). J. Am. Chem. Soc. 1970, 92, 2821–2825. [Google Scholar] [CrossRef]
- Miller, L.L.; Watkins, B.F. Scope and Mechanism of Aromatic Iodination with Electrochemically Generated Iodine(I). J. Am. Chem. Soc. 1976, 98, 1515–1519. [Google Scholar] [CrossRef]
- Midorikawa, K.; Suga, S.; Yoshida, J.-I. Selective monoiodination of aromatic compounds with electrochemically generated I+ using micromixing. Chem. Commun. 2006, 36, 3794–3796. [Google Scholar] [CrossRef]
- Kataoka, K.; Hagiwara, Y.; Midorikawa, K.; Suga, S.; Yoshida, J.-I. Practical electrochemical iodination of aromatic compounds. Org. Process. Res. Dev. 2008, 12, 1130–1136. [Google Scholar] [CrossRef]
- Yan, L.; Lei, H.; Yang, P.; Zhang, W. Electrochemically Generated Iodine Cations from a Glassy Carbon Electrode for Highly Selective Iodination of Anisole. Trans. Tianjin Univ. 2022, 28, 433–439. [Google Scholar] [CrossRef]
- Palmer, D.A.; Lietzke, M.H. The Equilibria and Kinetics of Iodine Hydrolysis. Radiochim. Acta 1982, 31, 37–44. [Google Scholar] [CrossRef]
- Paquette, J.; Ford, B.L. Ford, Iodine chemistry in the +1 oxidation state. I. The electronic spectra of OI−, HOI and H2OI+. Can. J. Chem. 1985, 63, 2444–2448. [Google Scholar] [CrossRef] [Green Version]
- Lengyel, I.; Epstein, I.R.; Kustin, K. Kinetics of Iodine Hydrolysis. Inorg. Chem. 1993, 32, 5880–5882. [Google Scholar] [CrossRef]
- Kolthoff, I.M.; Jordan, J. Voltammetry of Iodine and Iodide at Rotated Platinum Wire Electrodes. J. Am. Chem. Soc. 1953, 75, 1571–1575. [Google Scholar] [CrossRef]
- Miller, F.; Zittel, H. Voltammetry of the iodine system in aqueous medium at the pyrolytic graphite electrode. J. Electroanal. Chem. 1966, 11, 85–93. [Google Scholar] [CrossRef]
- Zittel, H.; Miller, F. Chronopotentiometry of the Iodine System at the Pyrolytic-Graphite and Glassy-Carbon Electrodes. J. Electroanal. Chem. 1967, 13, 193–207. [Google Scholar] [CrossRef]
- Dryhurst, G.; Elving, P.J. Electrooxidation of Halides at Pyrolytic Graphite Electrode in Aqueous and Acetonitrile Solutions. Anal. Chem. 1967, 39, 606–615. [Google Scholar] [CrossRef]
- Ito, S.; Sugimasa, M.; Toshimitsu, Y.; Orita, A.; Kitagawa, M.; Sakai, M. Anodic and cathodic modification of glassy-carbon electrodes affect iodine electrochemistry. Electrochim. Acta 2021, 379, 138181. [Google Scholar] [CrossRef]
- Pourbaix, M. Atlas of Electrochemical Equilibria in Aqueous Solutions, 2nd ed.; National Association of Corrosion Engineers: Huston, TX, USA, 1974. [Google Scholar]
- Varenikov, A.; Shapiro, E.; Gandelman, M. Decarboxylative Halogenation of Organic Compounds. Chem. Rev. 2020, 121, 412–484. [Google Scholar] [CrossRef]
- Chen, T.Q.; Pedersen, P.S.; Dow, N.W.; Fayad, R.; Hauke, C.E.; Rosko, M.C.; Danilov, E.O.; Blakemore, D.C.; Dechert-Schmitt, A.-M.; Knauber, T.; et al. A Unified Approach to Decarboxylative Halogenation of (Hetero)aryl Carboxylic Acids. J. Am. Chem. Soc. 2022, 144, 8296–8305. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sorti, L.; Vitulano, F.; Cappellini, E.; Uggeri, F.; Morelli, C.F.; Sello, G.; Minguzzi, A.; Vertova, A. Electrochemical Iodination through the In Situ Generation of Iodinating Agents: A Promising Green Approach. Molecules 2023, 28, 5555. https://doi.org/10.3390/molecules28145555
Sorti L, Vitulano F, Cappellini E, Uggeri F, Morelli CF, Sello G, Minguzzi A, Vertova A. Electrochemical Iodination through the In Situ Generation of Iodinating Agents: A Promising Green Approach. Molecules. 2023; 28(14):5555. https://doi.org/10.3390/molecules28145555
Chicago/Turabian StyleSorti, Letizia, Fiammetta Vitulano, Elia Cappellini, Fulvio Uggeri, Carlo Francesco Morelli, Guido Sello, Alessandro Minguzzi, and Alberto Vertova. 2023. "Electrochemical Iodination through the In Situ Generation of Iodinating Agents: A Promising Green Approach" Molecules 28, no. 14: 5555. https://doi.org/10.3390/molecules28145555
APA StyleSorti, L., Vitulano, F., Cappellini, E., Uggeri, F., Morelli, C. F., Sello, G., Minguzzi, A., & Vertova, A. (2023). Electrochemical Iodination through the In Situ Generation of Iodinating Agents: A Promising Green Approach. Molecules, 28(14), 5555. https://doi.org/10.3390/molecules28145555