
A Validated HPLC-UV-ESI-IT-MS Method for the Quantification of Carnosol in Lepechinia mutica, a Medicinal Plant Endemic to Ecuador

 , , , and
, , , and 
Abstract
:
1. Introduction
2. Results and Discussion
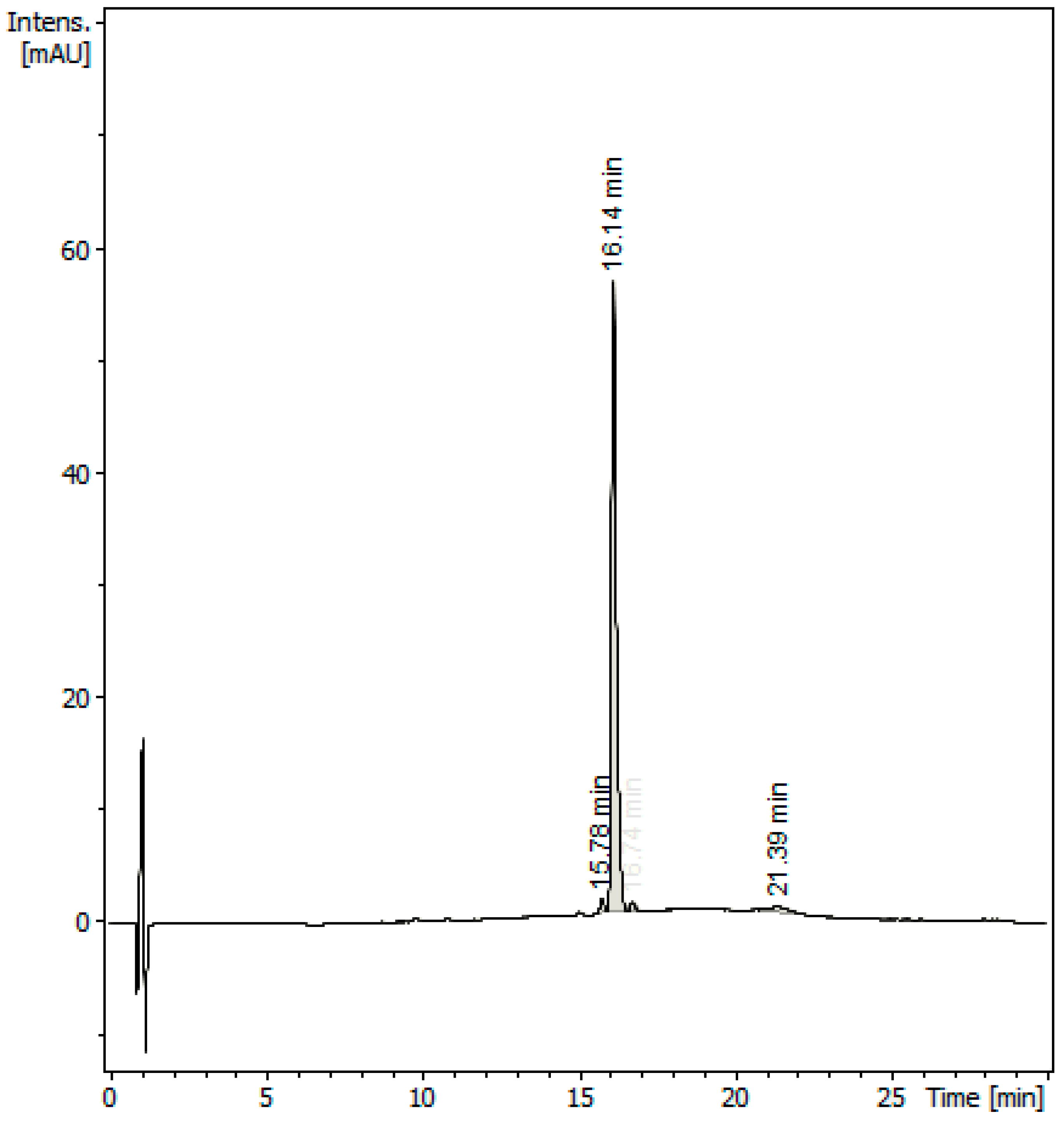
2.1. Specificity of the Method
2.2. Linearity Study of Standard Carnosol
2.3. Lowest Limit of Detection (LOD) and Lowest Limit of Quantitation (LOQ) of Standard Carnosol
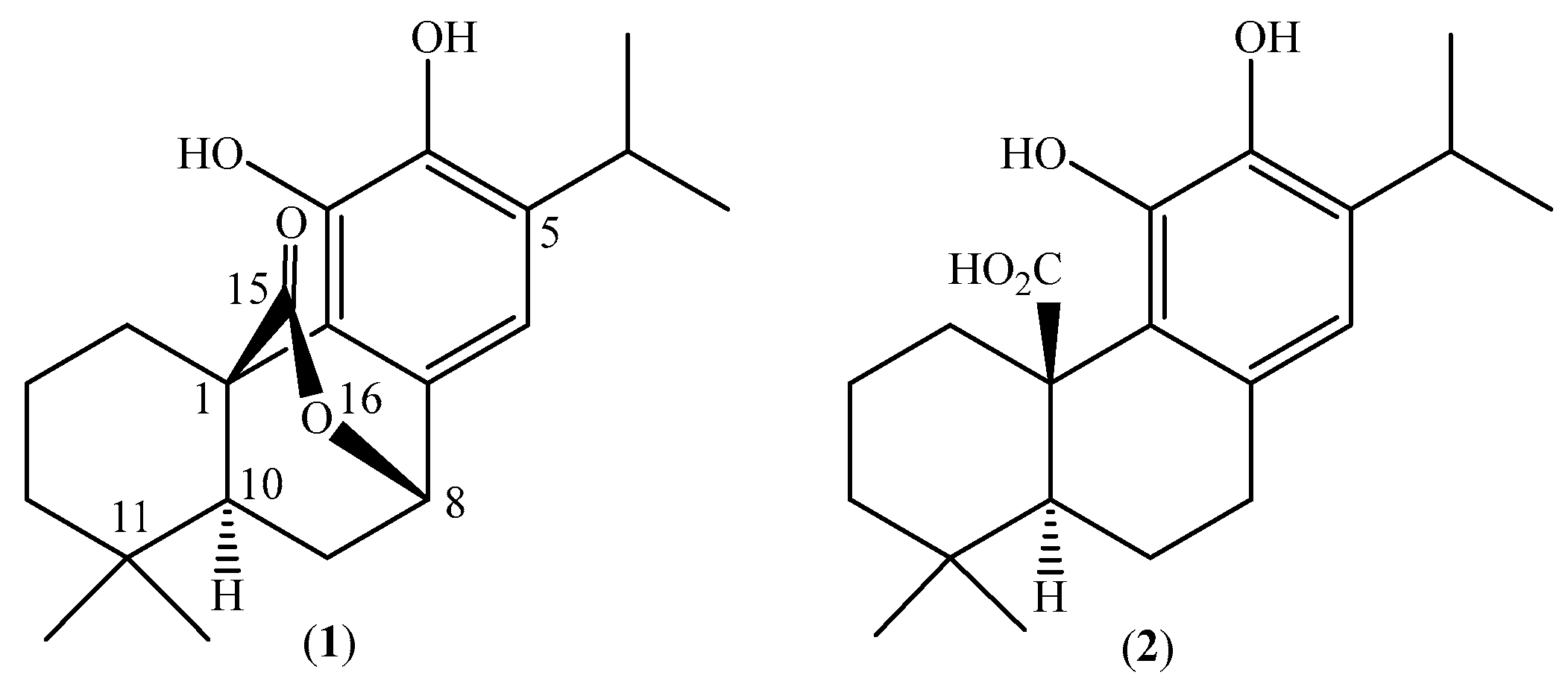
2.4. Quantification of Carnosol in Lepechinia mutica
2.5. Analytical Method Validation
2.5.1. Precision
Repeatability
Intermediate Precision
2.5.2. Accuracy (% Recovery)
2.5.3. Robustness
3. Materials and Methods
3.1. General Information
3.2. Plant Material
3.3. Standard Carnosol
3.4. HPLC Analysis of Standard Carnosol
3.5. MS Chromatographic Conditions
3.6. Linearity Range, Limit of Detection (LOD), and Limit of Quantitation (LOQ) of Standard Carnosol
3.7. Quantification of Carnosol in Lepechinia mutica Leaves
- C: Content (mg/g) of carnosol (1) in the dried leaves
- Co: Mean concentration of carnosol (1) in the dissolved sample of A’ = 43.1 · 10−3 mg/mL
- V: Volume (1.5 mL) of MeOH-H2O, 9:1 v/v, used to dissolve the sample of A’
- WA: Weight of total residue A’ = 50 · 103 mg
- WB: Weight of the sample of A’ from which chlorophyll was removed = 10 mg
- W: Weight of dried leaves = 400 g
3.8. Validation of the Analytical Method
3.8.1. Precision (Repeatability and Intermediate Precision)
3.8.2. Accuracy
3.8.3. Robustness
- R: Peak resolution
- Tr1: Retention time (min) of the compound that elutes first (rosmarinic acid)
- Tr2: Retention time (min) of the compound that elutes second (carnosol)
- W1: Peak width (min) of rosmarinic acid
- W2: Peak width (min) of carnosol
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Brieskorn, C.H.; Fuchs, A.; Bredenberg, J.B.-S.; McChesney, J.D.; Wenkert, E. The structure of carnosol. J. Org. Chem. 1964, 29, 2293–2298. [Google Scholar] [CrossRef]
- Birtić, S.; Dussort, P.; Pierre, F.-X.; Bily, A.C.; Roller, M. Carnosic acid. Phytochemistry 2015, 115, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, F.M.; Farimani, M.M.; Amin, G. Carnosol from Salvia eremophila Boiss. DARU 2000, 8, 45–46. [Google Scholar]
- Guerrero, I.C.; Andrés, L.S.; León, L.G.; Machín, R.P.; Padrón, J.M.; Luis, J.G.; Delgadillo, J. Abietane diterpenoids from Salvia pachyphylla and S. clevelandii with cytotoxic activity against human cancer cell lines. J. Nat. Prod. 2006, 69, 1803–1805. [Google Scholar] [CrossRef]
- Areche, C.; Schmeda-Hirschmann, G.; Theoduloz, C.; Rodríguez, J.A. Gastroprotective effect and cytotoxicity of abietane diterpenes from the Chilean Lamiaceae Sphacele chamaedryoides (Balbis) Briq. J. Pharm. Pharmacol. 2009, 61, 1689–1697. [Google Scholar] [CrossRef]
- Encarnación-Dimayuga, R.; Almada, G.; Virgen, M. Minimum antimicrobial inhibitory concentration of carnosol and of the ethanol extract from Lepichinia hastata (Lamiaceae). Phytomedicine 1998, 5, 301–305. [Google Scholar] [CrossRef]
- Ramírez, J.; Gilardoni, G.; Ramón, E.; Tosi, S.; Picco, A.M.; Bicchi, C.; Vidari, G. Phytochemical study of the Ecuadorian species Lepechinia mutica (Benth.) Epling and high antifungal activity of carnosol against Pyricularia oryzae. Pharmaceuticals 2018, 11, 33. [Google Scholar] [CrossRef]
- Marrero, J.G.; Moujir, L.; Andrés, L.S.; Montaño, N.P.; Araujo, L.; Luis, J.G. Semisynthesis and biological evaluation of abietane-type diterpenes. Revision of the structure of rosmaquinone. J. Nat. Prod. 2009, 72, 1385–1389. [Google Scholar] [CrossRef]
- Núñez, M.J.; Reyes, C.P.; Jiménez, I.A.; Hayashi, H.; Tokuda, H.; Bazzocchi, I.L. ent-Rosane and abietane diterpenoids as cancer chemopreventive agents. Phytochemistry 2011, 72, 385–390. [Google Scholar] [CrossRef]
- Johnson, J.J. Carnosol: A promising anti-cancer and anti-inflammatory agent. Cancer Lett. 2011, 305, 1–7. [Google Scholar] [CrossRef]
- Bauer, J.; Kuehnl, S.; Rollinger, J.M.; Scherer, O.; Northoff, H.; Stuppner, H.; Werz, O.; Koeberle, A. Carnosol and carnosic acids from Salvia officinalis inhibit microsomal prostaglandin E2 synthase-1. J. Pharmacol. Exp. Ther. 2012, 342, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Ceylan, B.; Tiris, G.; Tekkeli, S.E.K. A new HPLC method with UV detection for the determination of carnosol in human plasma and application to a pharmacokinetic study. Chromatographia 2021, 84, 855–860. [Google Scholar] [CrossRef]
- Ramírez, J.; Suarez, A.I.; Bec, N.; Armijos, C.; Gilardoni, G.; Larroque, C.; Vidari, G. Carnosol from Lepechinia mutica and tiliroside from Vallea stipularis: Two promising inhibitors of BuChE. Rev. Bras. Farmacogn. 2018, 28, 559–563. [Google Scholar] [CrossRef]
- Gilardoni, G.; Ramírez, J.; Montalván, M.; Quinche, W.; León, J.; Benítez, L.; Morocho, V.; Cumbicus, N.; Bicchi, C. Phytochemistry of three Ecuadorian Lamiaceae: Lepechinia heteromorpha (Briq.) Epling, Lepechinia radula (Benth.) Epling and Lepechinia paniculata (Kunth) Epling. Plants 2019, 8, 1. [Google Scholar] [CrossRef]
- Ramírez, J.; Gilardoni, G.; Jácome, M.; Montesinos, J.; Rodolfi, M.; Guglielminetti, M.L.; Cagliero, C.; Bicchi, C.; Vidari, G. Chemical composition, enantiomeric analysis, AEDA sensorial evaluation and antifungal activity of the essential oil from the Ecuadorian plant Lepechinia mutica Benth (Lamiaceae). Chem. Biodivers. 2017, 14, e1700292. [Google Scholar] [CrossRef]
- Jørgensen, P.M.; León-Yánez, S. Catalogue of the Vascular Plants of Ecuador; Missouri Botanical Garden Press: St. Louis, MO, USA, 1999; Volume 75. [Google Scholar]
- Drew, B.T.; Sytsma, K.J. The South American radiation of Lepechinia (Lamiaceae): Phylogenetics, divergence times and evolution of dioecy. Bot. J. Linn. Soc. 2013, 171, 171–190. [Google Scholar] [CrossRef]
- Naranjo, P.; Escaleras, R. Medicina Tradicional en el Ecuador; Corporación Editorial Nacional: Quito, Ecuador, 1995. [Google Scholar]
- Tene, V.; Malagón, O.; Finzi, P.V.; Vidari, G.; Armijos, C.; Zaragoza, T. An ethnobotanical survey of medicinal plants used in Loja and Zamora-Chinchipe, Ecuador. J. Ethnopharmacol. 2007, 111, 63–81. [Google Scholar] [CrossRef]
- Armijos, C.; Ramírez, J.; Salinas, M.; Vidari, G.; Suárez, A.I. Pharmacology and phytochemistry of Ecuadorian medicinal plants: An update and perspectives. Pharmaceuticals 2021, 14, 1145. [Google Scholar] [CrossRef]
- Reuhs, B.L. High-Performance Liquid Chromatography. In Food Analysis; Nielsen, S.S., Ed.; Food Science Text Series; Springer: Cham, Switzerland, 2017. [Google Scholar] [CrossRef]
- Gajhede, M.; Anthoni, U.; Per Nielsen, H.; Pedersen, E.J.; Christophersen, C. Carnosol. Crystal structure, absolute configuration, and spectroscopic properties of a diterpene. J. Crystallogr. Spectrosc. Res. 1990, 20, 165–171. [Google Scholar] [CrossRef]
- Thompson, M.; Ellison, S.L.R.; Wood, R. Harmonized guidelines for single-laboratory validation of methods of analysis (IUPAC Technical Report). Pure Appl. Chem. 2002, 74, 835–855. [Google Scholar] [CrossRef]
- Validation of Analytical Procedures: Text and Methodology (Q2(R1)). In Proceedings of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use, London, UK, November 2005.
- Magnusson, B.; Örnemark, U. Eurachem Guide—The Fitness for Purpose of Analytical Methods–A Laboratory Guide to Method Validation and Related Topics, 2nd ed.; LGC: Teddington, UK, 2014. [Google Scholar]
- Sabir, A.M.; Moloy, M.; Bhasin, P.S. HPLC method development and validation: A review. Int. Res. J. Pharm. 2013, 4, 39–46. [Google Scholar] [CrossRef]
- Singtoroj, T.; Tarning, J.; Annerberg, A.; Ashton, M.; Bergqvist, Y.; White, N.J.; Lindegardh, N.; Day, N.P.J. A new approach to evaluate regression models during validation of bioanalytical assays. J. Pharm. Biomed. Anal. 2006, 41, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Raposo, F. Evaluation of analytical calibration based on least-squares linear regression for instrumental techniques: A tutorial review. TrAC Trends Anal. Chem. 2016, 77, 167–185. [Google Scholar] [CrossRef]
- AOAC Official Methods of Analysis. Appendix F: Guidelines for Standard Method Performance Requirements; AOAC International: Washington, DC, USA, 2016; pp. 1–18. [Google Scholar]
- University of Tartu. LC-MS Method Validation. 10.3 Different Ways to Evaluate Robustness|MOOC: Validation of Liquid Chromatography Mass Spectrometry (LC-MS) Methods (Analytical Chemistry) Course (ut.ee). Available online: https://sisu.ut.ee/lcms_method_validation/103-different-ways-evaluate-robustness (accessed on 25 July 2023).
- Loussouarn, M.; Krieger-Liszkay, A.; Svilar, L.; Bily, A.; Birtić, S.; Havaux, M. Carnosic acid and carnosol, two major antioxidants of rosemary, act through different mechanisms. Plant Physiol. 2017, 175, 1381–1394. [Google Scholar] [CrossRef] [PubMed]
- Akihisa, T.; Yasukawa, K.; Tokuda, H. Potentially cancer chemopreventive and anti-inflammatory terpenoids from natural sources. In Studies in Natural Products Chemistry; Rahman, A.U., Ed.; Elsevier: Amsterdam, The Netherlands, 2003; Volume 29, Part J; pp. 73–126. [Google Scholar]
- Troncoso, N.; Sierra, H.; Carvajal, L.; Delpiano, P.; Günther, G. Fast high performance liquid chromatography and ultraviolet-visible quantification of principal phenolic antioxidants in fresh rosemary. J. Chromatogr. A 2005, 1100, 20–25. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Resolution | RSD, % | |
|---|---|---|---|
| Flow (mL/min) | 0.2 | 0.77 ± 0.05 | 4.5 |
| 0.4 | 0.74 ± 0.01 | ||
| UV detection wavelength (nm) | 278 | 0.78 ± 0.01 | 2.7 |
| 283 | 0.77 ± 0.00 | ||
| 290 | 0.78 ± 0.04 | ||
| Oven temperature (°C) | 18 | 0.82 ± 0.01 | 4.6 |
| 20 | 0.81 ± 0.01 | ||
| 25 | 0.79 ± 0.07 | ||
| Mobile phase composition a | A: 27/73/0.1; B: 56/44/0.1 | 0.70 ± 0.04 | 4.9 |
| A: 29/71/01; B: 58/42/01 | 0.74 ± 0.02 | ||
| A: 31/69/01; B: 62/38/01 | 0.70 ± 0.03 | ||
| Gradient program b | 1 | 0.76 ± 0.03 | 3.6 |
| 2 | 0.79 ± 0.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solano-Cueva, N.; Figueroa, J.G.; Loja, C.; Armijos, C.; Vidari, G.; Ramírez, J. A Validated HPLC-UV-ESI-IT-MS Method for the Quantification of Carnosol in Lepechinia mutica, a Medicinal Plant Endemic to Ecuador. Molecules 2023, 28, 6701. https://doi.org/10.3390/molecules28186701
Solano-Cueva N, Figueroa JG, Loja C, Armijos C, Vidari G, Ramírez J. A Validated HPLC-UV-ESI-IT-MS Method for the Quantification of Carnosol in Lepechinia mutica, a Medicinal Plant Endemic to Ecuador. Molecules. 2023; 28(18):6701. https://doi.org/10.3390/molecules28186701
Chicago/Turabian StyleSolano-Cueva, Natalí, Jorge G. Figueroa, Corina Loja, Chabaco Armijos, Giovanni Vidari, and Jorge Ramírez. 2023. "A Validated HPLC-UV-ESI-IT-MS Method for the Quantification of Carnosol in Lepechinia mutica, a Medicinal Plant Endemic to Ecuador" Molecules 28, no. 18: 6701. https://doi.org/10.3390/molecules28186701
APA StyleSolano-Cueva, N., Figueroa, J. G., Loja, C., Armijos, C., Vidari, G., & Ramírez, J. (2023). A Validated HPLC-UV-ESI-IT-MS Method for the Quantification of Carnosol in Lepechinia mutica, a Medicinal Plant Endemic to Ecuador. Molecules, 28(18), 6701. https://doi.org/10.3390/molecules28186701