
Explaining Accurate Predictions of Multitarget Compounds with Machine Learning Models Derived for Individual Targets



Abstract
:1. Introduction
2. Results and Discussion
2.1. Compound Test System and Random Forest Models
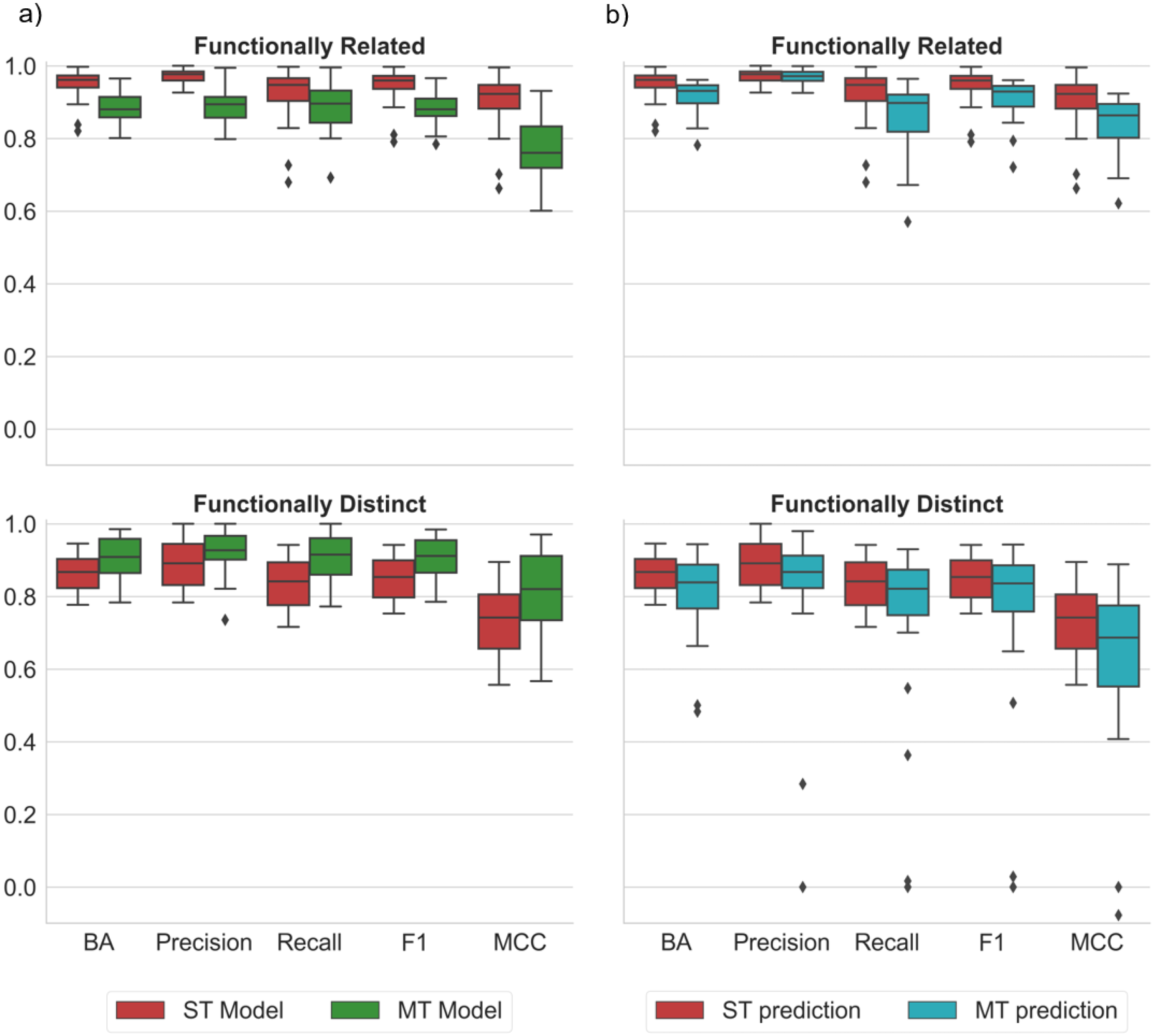
2.2. ST and MT Model Performance
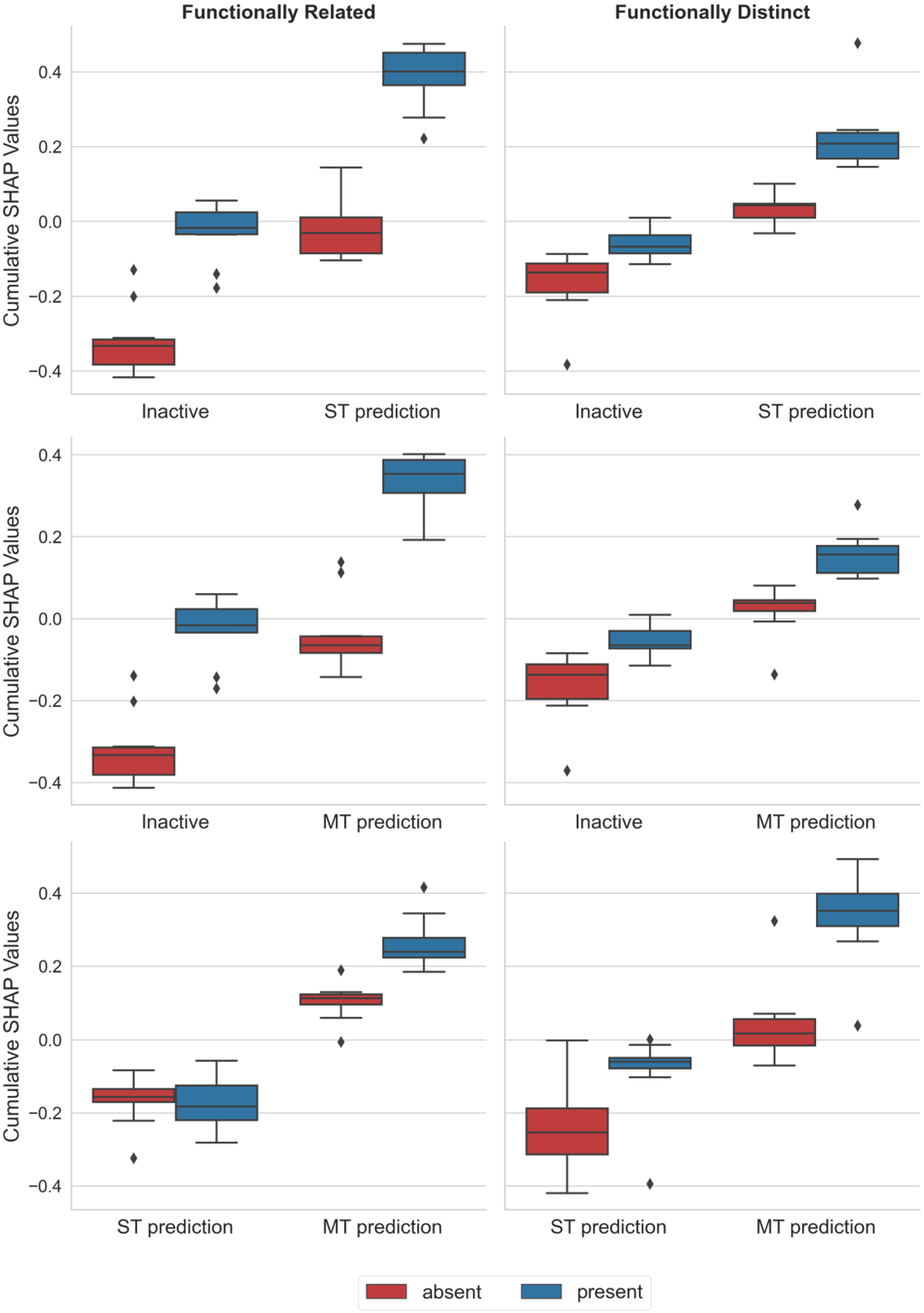
2.3. Rationalizing Model Decisions
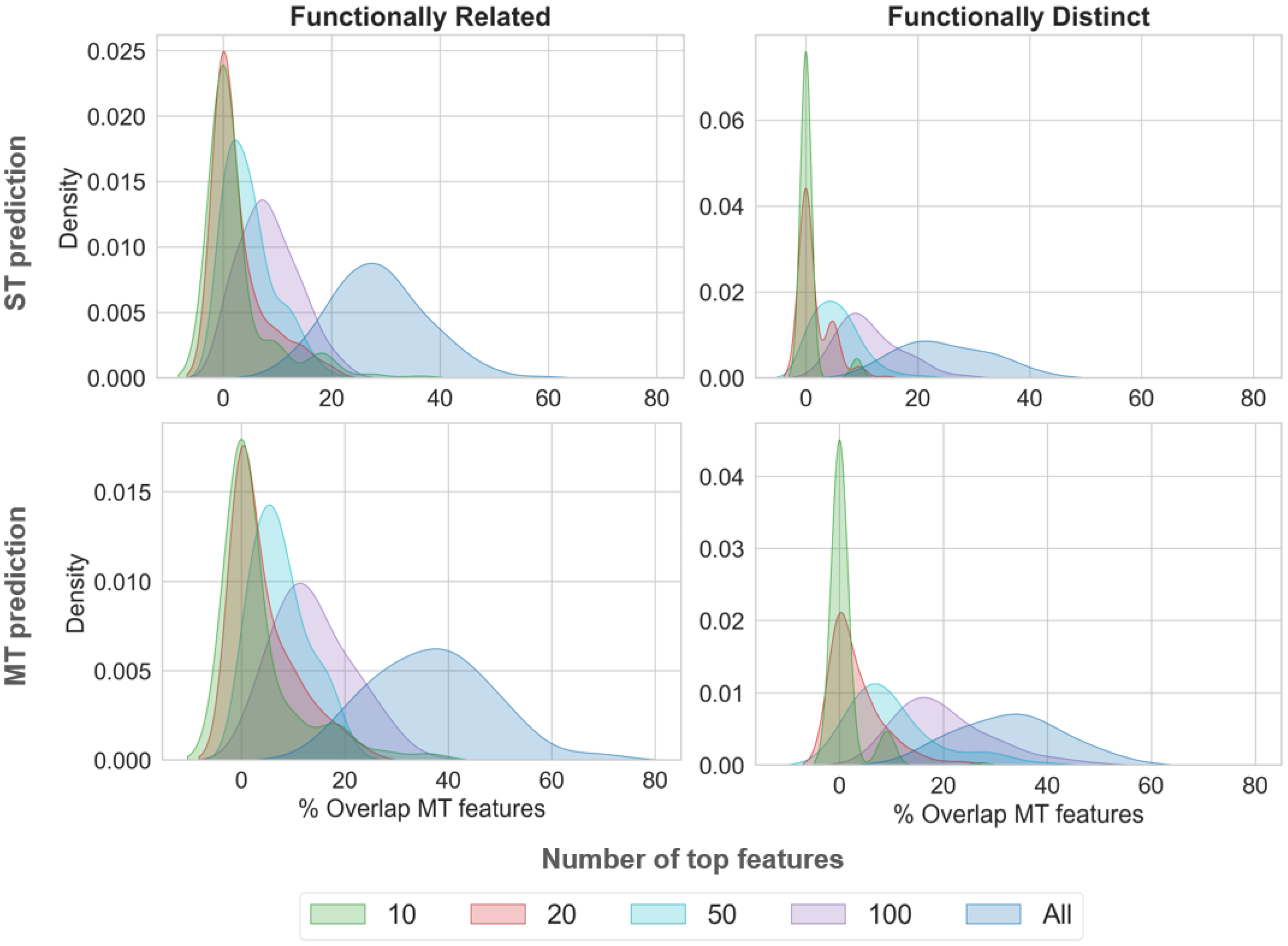
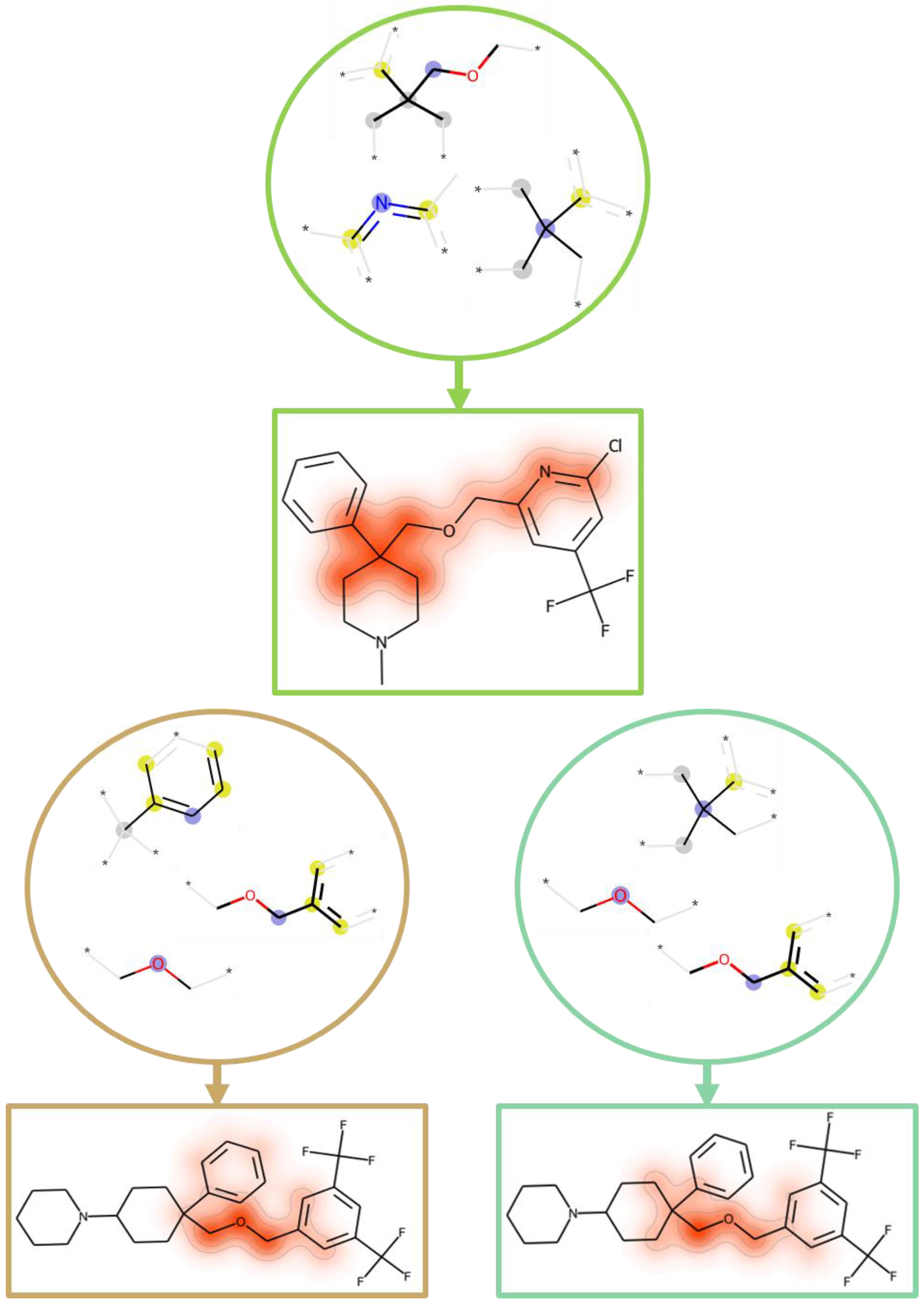
2.4. Comparative Feature Analysis
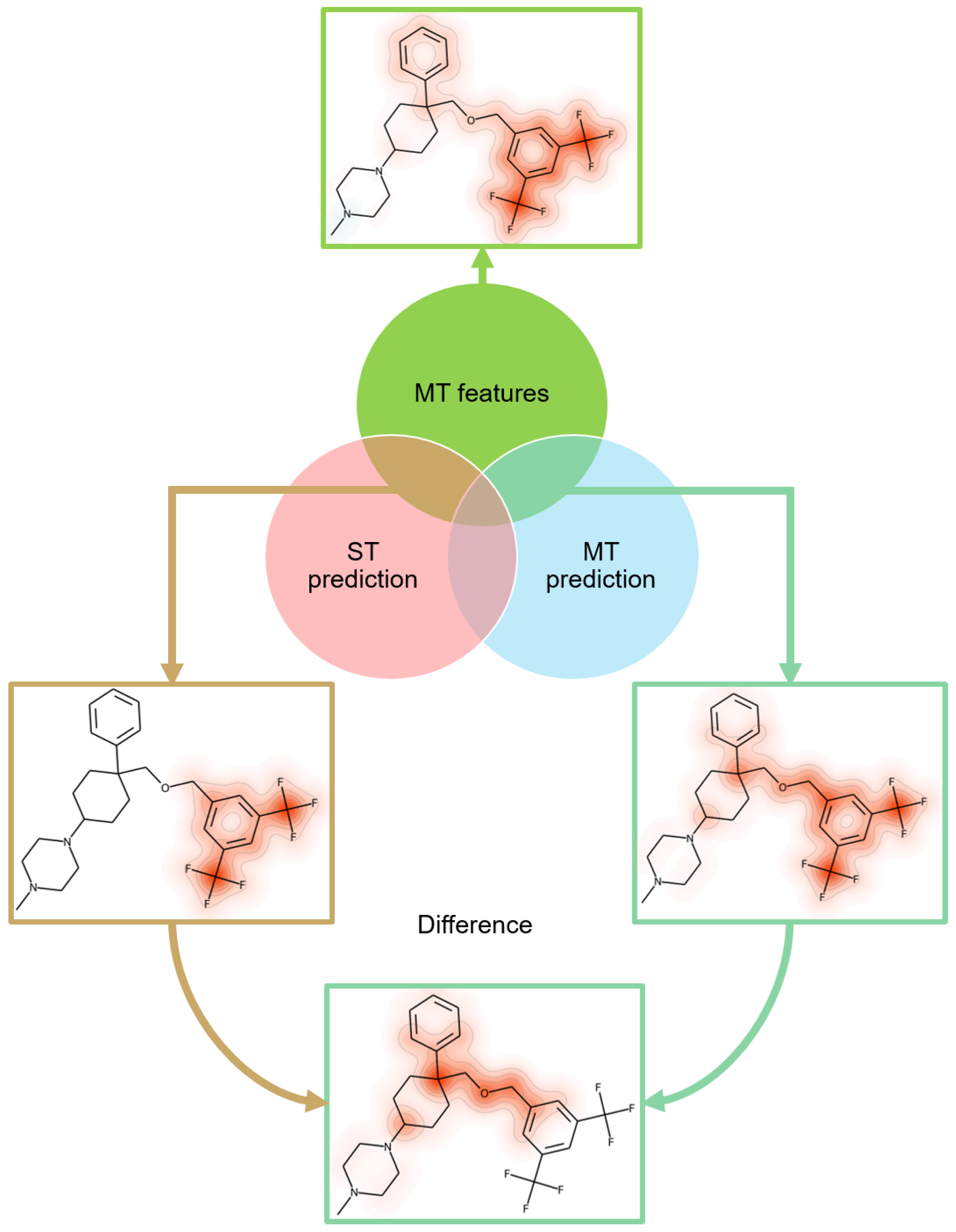
2.5. Feature Mapping
2.6. Conclusions
3. Materials and Methods
3.1. Compound Activity Classes and Target Pairs
3.2. Molecular Representation
3.3. Machine Learning
3.3.1. Model Training, Hyperparameter Optimization, and Predictions
3.3.2. Performance Measures
3.4. Model Explanation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Bolognesi, M.L.; Cavalli, A. Multitarget Drug Discovery and Polypharmacology. ChemMedChem 2016, 11, 1190–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Bajorath, J. Compound Promiscuity—What Can We Learn from Current Data. Drug Discov. Today 2013, 18, 644–650. [Google Scholar] [CrossRef]
- Feldmann, C.; Miljković, F.; Yonchev, D.; Bajorath, J. Identifying Promiscuous Compounds with Activity against Different Target Classes. Molecules 2019, 24, 4185. [Google Scholar] [CrossRef] [Green Version]
- Bolognesi, L.M. Polypharmacology in a Single Drug: Multitarget Drugs. Curr. Med. Chem. 2013, 20, 1639–1645. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and Opportunities in Drug Discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2019, 62, 420–444. [Google Scholar] [CrossRef]
- Rastelli, G.; Pinzi, L. Computational Polypharmacology Comes of Age. Front. Pharmacol. 2015, 6, 157. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Jiang, X.; He, S.; Jiang, H.; Feng, F.; Liu, W.; Qu, W.; Sun, H. Rational Design of Multitarget-Directed Ligands: Strategies and Emerging Paradigms. J. Med. Chem. 2019, 62, 8881–8914. [Google Scholar] [CrossRef]
- Chaudhari, R.; Fong, L.W.; Tan, Z.; Huang, B.; Zhang, S. An Up-To-Date Overview of Computational Polypharmacology in Modern Drug Discovery. Expert Opin. Drug Discov. 2020, 15, 1025–1044. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, C.; Yonchev, D.; Stumpfe, D.; Bajorath, J. Systematic Data Analysis and Diagnostic Machine Learning Reveal differences between Compounds with Single- and Multitarget Activity. Mol. Pharm. 2020, 17, 4652–4666. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, C.; Yonchev, D.; Bajorath, J. Analysis of Biological Screening Compounds with Single- or Multi-Target Activity via Diagnostic Machine Learning. Biomolecules 2020, 10, 1605. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, C.; Bajorath, J. Machine Learning Reveals that Structural Features Distinguishing Promiscuous and Non-Promiscuous Compounds Depend on Target Combinations. Sci. Rep. 2021, 11, 7863. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.; Bento, A.P.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrián-Uhalte, E.; et al. The ChEMBL Database in 2017. Nucleic Acids Res. 2017, 45, 945–954. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Duan, D.; Torosyan, H.; Doak, A.K.; Ziebart, K.T.; Sterling, T.; Tumanian, G.; Shoichet, B.K. An Aggregation Advisor for Ligand Discovery. J. Med. Chem. 2015, 58, 1712–1722. [Google Scholar] [CrossRef] [Green Version]
- RDKit: Cheminformatics and Machine Learning Software. 2013. Available online: http://www.rdkit.org (accessed on 15 November 2022).
- Bruns, R.F.; Watson, I.A. Rules for Identifying Potentially Reactive or Promiscuous Compounds. J. Med. Chem. 2012, 55, 9763–9772. [Google Scholar] [CrossRef]
- UniProt: The Universal Protein Knowledgebase in 2021. Nucleic Acids Res. 2021, 49, 480–489. Available online: https://www.uniprot.org/ (accessed on 1 December 2022). [CrossRef] [PubMed]
- Rogers, D.; Hahn, M. Extended-Connectivity Fingerprints. J. Chem. Inf. Model. 2010, 50, 742–754. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Brodersen, K.H.; Ong, C.S.; Stephan, K.E.; Buhmann, J.M. The balanced accuracy and its posterior distribution. In Proceedings of the 20th International Conference on Pattern Recognition (ICPR), Istanbul, Turkey, 23–26 August 2010; pp. 3121–3124. [Google Scholar]
- Van Rijsbergen, C.J. Information Retrieval, 2nd ed.; Butterworth-Heinemann: Oxford, UK, 1979. [Google Scholar]
- Matthews, B.W. Comparison of the Predicted and Observed Secondary Structure of T4 Phage Lysozyme. BBA Protein Struct. 1975, 405, 442–451. [Google Scholar] [CrossRef]
- Shapley, L.S. A value for N-Person games. In Contributions to the Theory of Games. Annals of Mathematical Studies; Kuhn, H.W., Tucker, A.W., Eds.; Princeton University Press: Princeton, NJ, USA, 1953; pp. 307–317. [Google Scholar]
- Lundberg, S.M.; Lee, S. A unified approach to interpreting model predictions. In Proceedings of the 31st International Conference on Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017; pp. 4768–4777. [Google Scholar]
- Rodríguez-Pérez, R.; Bajorath, J. Interpretation of Compound Activity Predictions from Complex Machine Learning Models Using Local Approximations and Shapley Values. J. Med. Chem. 2019, 63, 8761–8777. [Google Scholar] [CrossRef]
- Lundberg, S.M.; Erion, G.; Chen, H.; DeGrave, A.; Prutkin, J.M.; Nair, B.; Katz, R.; Himmelfarb, J.; Bansal, N.; Lee, S. From Local Explanations to Global Understanding with Explainable AI for Trees. Nat. Mach. Intell. 2020, 2, 56–67. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Functionally Related | Functionally Distinct |
|---|---|
| Sodium/glucose cotransporter 1 Sodium/glucose cotransporter 2 | Tumor necrosis factor Nucleotide-binding oligomerization domain-containing protein 1 |
| Carbonic anhydrase 4 Carbonic anhydrase 7 | 5-hydroxytryptamine receptor Sodium-dependent noradrenaline transporter |
| Carbonic anhydrase 4 Carbonic anhydrase 9 | D2 dopamine receptor Sodium-dependent serotonin transporter |
| Cathepsin B Cathepsin S | Acetylcholinesterase Amine oxidase B |
| Insulin-like growth factor 1 receptor kinase ALK receptor tyrosine kinase | Acetylcholinesterase Beta-secretase 1 |
| Histone deacetylase 2 Histone deacetylase 6 | Substance-P receptor Sodium-dependent serotonin transporter |
| Histone deacetylase 3 Histone deacetylase 8 | Amine oxidase B Adenosine receptor A2a |
| Coagulation factor X Plasminogen | D3 dopamine receptor Sodium-dependent serotonin transporter |
| Prothrombin Coagulation factor VII | Histamine H3 receptor Sodium-dependent serotonin transporter |
| Intestinal collagenase Collagenase 3 | Histamine H1 receptor Sodium-dependent serotonin transporter |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lamens, A.; Bajorath, J. Explaining Accurate Predictions of Multitarget Compounds with Machine Learning Models Derived for Individual Targets. Molecules 2023, 28, 825. https://doi.org/10.3390/molecules28020825
Lamens A, Bajorath J. Explaining Accurate Predictions of Multitarget Compounds with Machine Learning Models Derived for Individual Targets. Molecules. 2023; 28(2):825. https://doi.org/10.3390/molecules28020825
Chicago/Turabian StyleLamens, Alec, and Jürgen Bajorath. 2023. "Explaining Accurate Predictions of Multitarget Compounds with Machine Learning Models Derived for Individual Targets" Molecules 28, no. 2: 825. https://doi.org/10.3390/molecules28020825
APA StyleLamens, A., & Bajorath, J. (2023). Explaining Accurate Predictions of Multitarget Compounds with Machine Learning Models Derived for Individual Targets. Molecules, 28(2), 825. https://doi.org/10.3390/molecules28020825