
Novel 9-Benzylaminoacridine Derivatives as Dual Inhibitors of Phosphodiesterase 5 and Topoisomerase II for the Treatment of Colon Cancer
 ,
,  , and
, and
Abstract
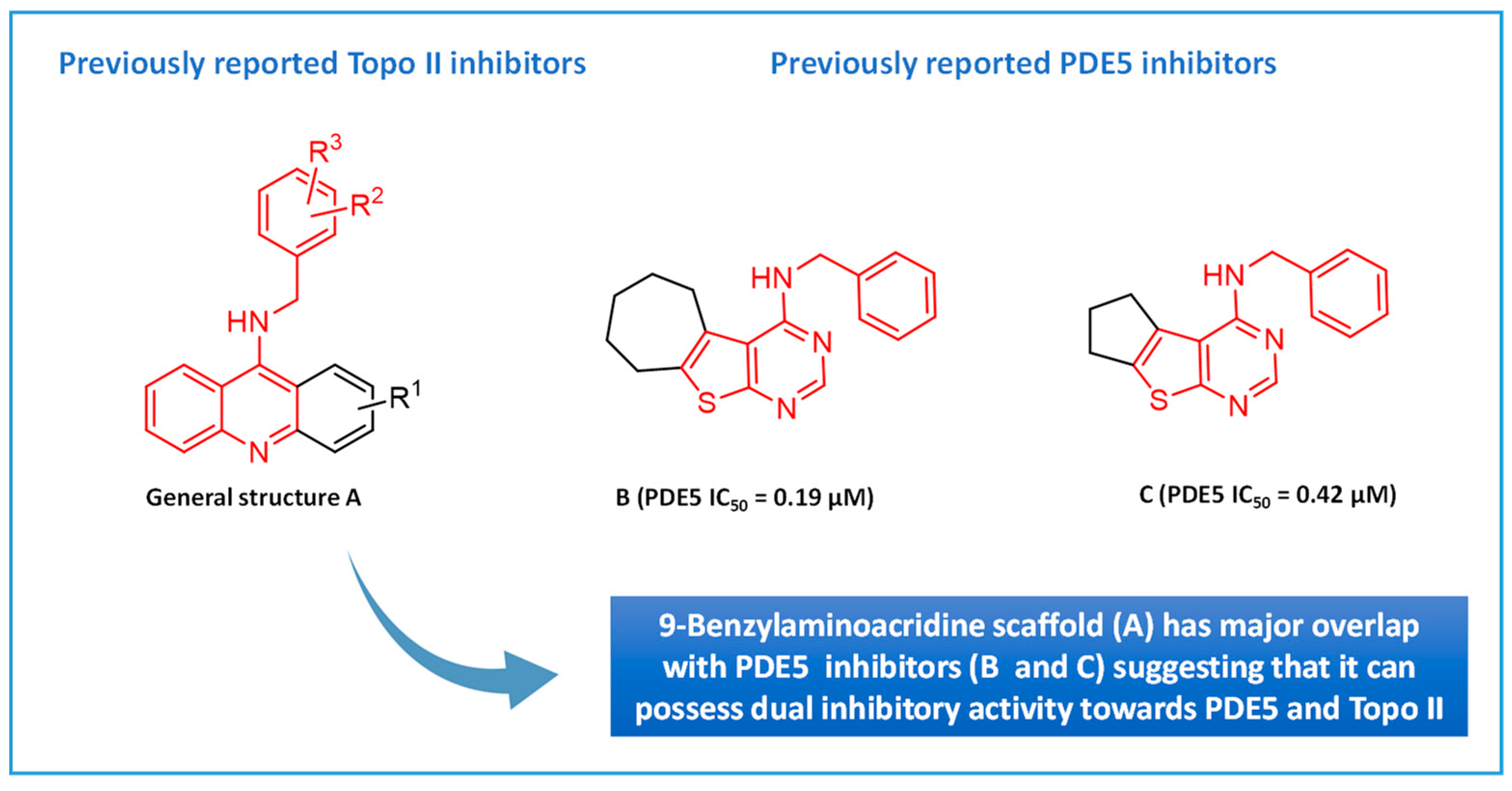
:1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Activity against PDE5
2.2.2. Testing the Anticancer Activity of the Compounds
NCI 60-Cell Line Testing
Effects of Compounds on Proliferation of Human Colorectal Cancer Cells and Non-Cancer Cells
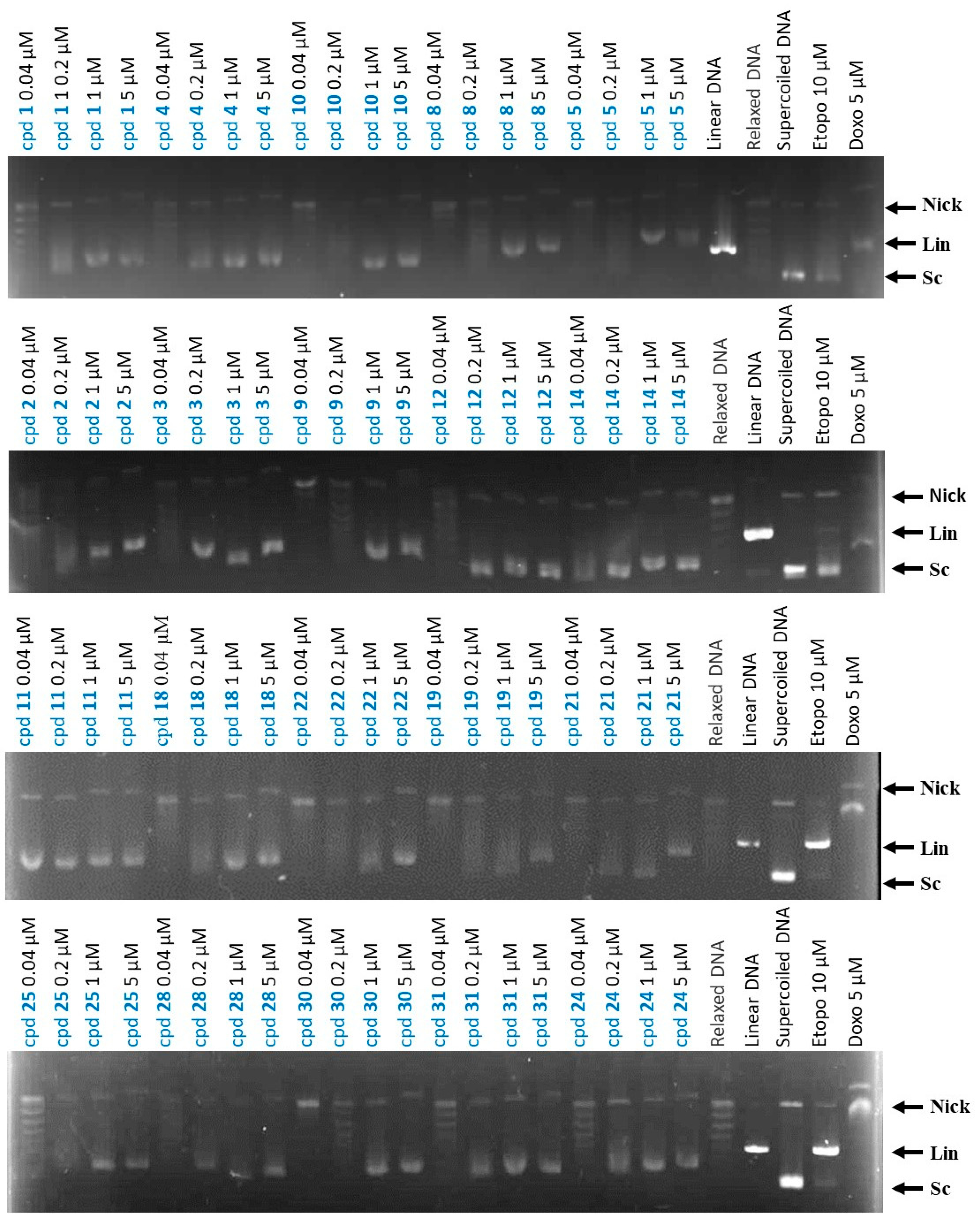
2.2.3. Effects of Compounds on Topo II Activity
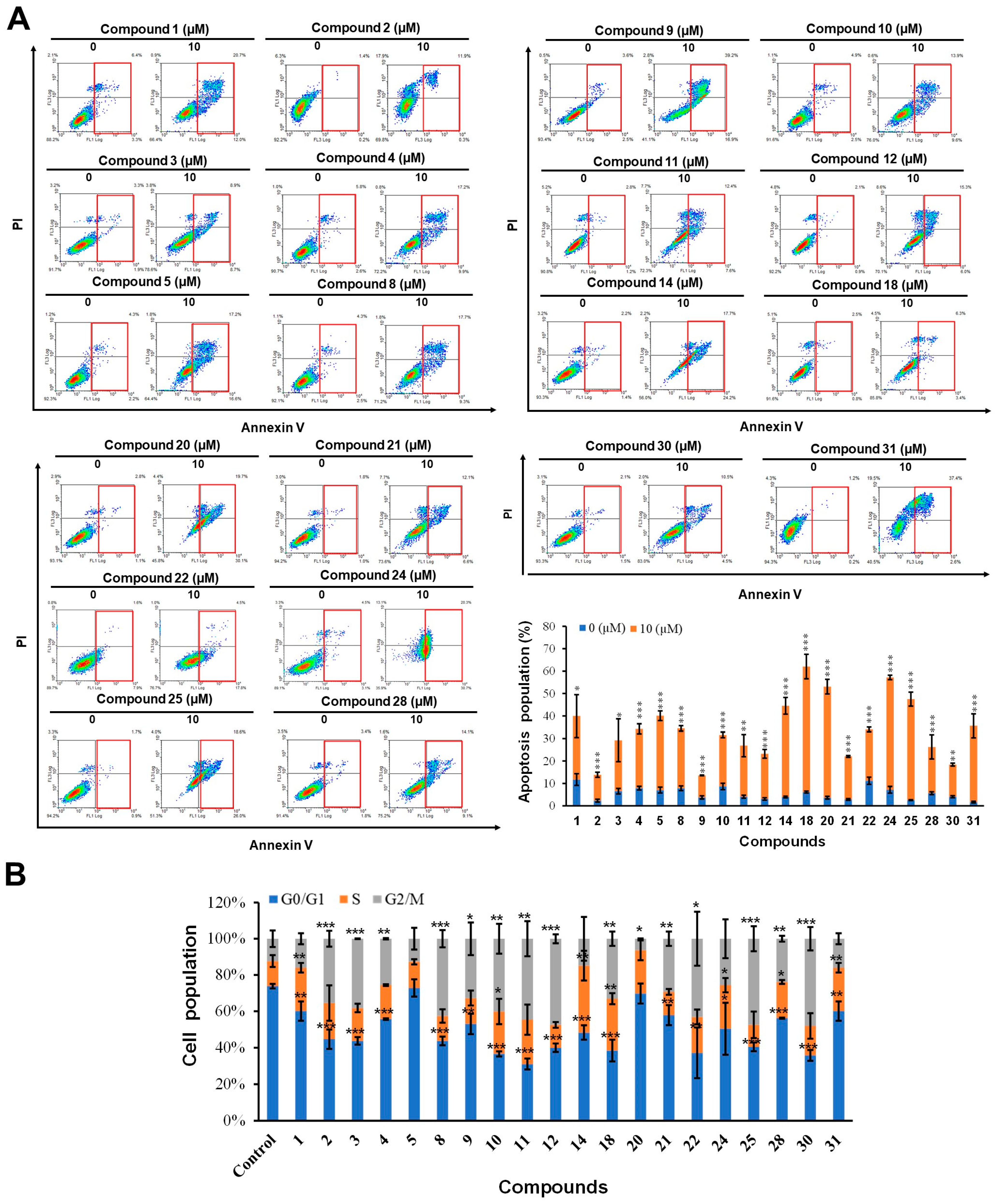
2.2.4. The Effects of Compounds on Apoptosis and Cell Cycle in HCT-116 Cells
3. Experimental Section
3.1. Chemistry
3.1.1. General Synthetic Procedures
Procedure A: General Procedure for Preparation of 2-Anilinobenzoic Acid Derivatives (A1–A3)
Procedure B: General Procedure for Synthesis of Acridone Derivatives (B1–B3)
Procedure C: General Procedure for Synthesis of 9-Chloroacridine Derivatives (C1–C3)
Procedure D: General Procedure for Synthesis of 9-Benzylacridine Analogs (1–31)
3.2. Biology
3.2.1. Cell Lines, Biomaterials, and Chemicals
3.2.2. Cell Proliferation Activity Assay
3.2.3. Apoptosis Assay
3.2.4. Cell Cycle Arrest Analysis
3.2.5. Determination of Topoisomerase II Inhibition
3.2.6. Phosphodiesterase 5 Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Corbin, J.; Francis, S. Pharmacology of phosphodiesterase-5 inhibitors. Int. J. Clin. Pract. 2002, 56, 453–459. [Google Scholar]
- Barnes, H.; Brown, Z.; Burns, A.; Williams, T. Phosphodiesterase 5 inhibitors for pulmonary hypertension. Cochrane Database Syst. Rev. 2019. [Google Scholar] [CrossRef]
- Rosen, R.C.; Kostis, J.B. Overview of phosphodiesterase 5 inhibition in erectile dysfunction. Am. J. Cardiol. 2003, 92, 9–18. [Google Scholar] [CrossRef]
- Rotella, D.P. Phosphodiesterase 5 inhibitors: Current status and potential applications. Nat. Rev. Drug Discov. 2002, 1, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Limin, M.; Johnsen, N.; Hellstrom, W.J. Avanafil, a new rapid-onset phosphodiesterase 5 inhibitor for the treatment of erectile dysfunction. Expert Opin. Investig. Drugs 2010, 19, 1427–1437. [Google Scholar] [CrossRef]
- Goldstein, I.; Burnett, A.L.; Rosen, R.C.; Park, P.W.; Stecher, V.J. The serendipitous story of sildenafil: An unexpected oral therapy for erectile dysfunction. Sex. Med. Rev. 2019, 7, 115–128. [Google Scholar] [CrossRef]
- Porst, H. IC351 (tadalafil, Cialis): Update on clinical experience. Int. J. Impot. Res. 2002, 14, S57–S64. [Google Scholar] [CrossRef]
- Keating, G.M.; Scott, L.J. Vardenafil. Drugs 2003, 63, 2673–2702. [Google Scholar] [CrossRef]
- Abdel-Halim, M.; Sigler, S.; Racheed, N.A.; Hefnawy, A.; Fathalla, R.K.; Hammam, M.A.; Maher, A.; Maxuitenko, Y.; Keeton, A.B.; Hartmann, R.W. From celecoxib to a novel class of phosphodiesterase 5 inhibitors: Trisubstituted pyrazolines as novel phosphodiesterase 5 inhibitors with extremely high potency and phosphodiesterase isozyme selectivity. J. Med. Chem. 2021, 64, 4462–4477. [Google Scholar] [CrossRef]
- Abdel-Halim, M.; Tinsley, H.; Keeton, A.B.; Weam, M.; Atta, N.H.; Hammam, M.A.; Hefnawy, A.; Hartmann, R.W.; Engel, M.; Piazza, G.A. Discovery of trisubstituted pyrazolines as a novel scaffold for the development of selective phosphodiesterase 5 inhibitors. Bioorganic Chem. 2020, 104, 104322. [Google Scholar] [CrossRef]
- Zhang, T.; Lai, Z.; Yuan, S.; Huang, Y.-Y.; Dong, G.; Sheng, C.; Ke, H.; Luo, H.-B. Discovery of evodiamine derivatives as highly selective PDE5 inhibitors targeting a unique allosteric pocket. J. Med. Chem. 2020, 63, 9828–9837. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, H.N.; Gary, B.D.; Keeton, A.B.; Zhang, W.; Abadi, A.H.; Reynolds, R.C.; Piazza, G.A. Sulindac sulfide selectively inhibits growth and induces apoptosis of human breast tumor cells by phosphodiesterase 5 inhibition, elevation of cyclic GMP, and activation of protein kinase G. Mol. Cancer Ther. 2009, 8, 3331–3340. [Google Scholar] [CrossRef] [Green Version]
- Fajardo, A.M.; Piazza, G.A.; Tinsley, H.N. The role of cyclic nucleotide signaling pathways in cancer: Targets for prevention and treatment. Cancers 2014, 6, 436–458. [Google Scholar] [CrossRef] [Green Version]
- Piazza, G.A.; Ward, A.; Chen, X.; Maxuitenko, Y.; Coley, A.; Aboelella, N.S.; Buchsbaum, D.J.; Boyd, M.R.; Keeton, A.B.; Zhou, G. PDE5 and PDE10 inhibition activates cGMP/PKG signaling to block Wnt/β-catenin transcription, cancer cell growth, and tumor immunity. Drug Discov. Today 2020, 25, 1521–1527. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Strada, S.J. The novel functions of cGMP-specific phosphodiesterase 5 and its inhibitors in carcinoma cells and pulmonary/cardiovascular vessels. Curr. Top. Med. Chem. 2007, 7, 437–454. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Vemavarapu, L.; Thompson, W.J.; Strada, S.J. Suppression of cyclic GMP-specific phosphodiesterase 5 promotes apoptosis and inhibits growth in HT29 cells. J. Cell. Biochem. 2005, 94, 336–350. [Google Scholar] [CrossRef]
- Mei, X.-L.; Yang, Y.; Zhang, Y.-J.; Li, Y.; Zhao, J.-M.; Qiu, J.-G.; Zhang, W.-J.; Jiang, Q.-W.; Xue, Y.-Q.; Zheng, D.-W. Sildenafil inhibits the growth of human colorectal cancer in vitro and in vivo. Am. J. Cancer Res. 2015, 5, 3311. [Google Scholar]
- Pantziarka, P.; Sukhatme, V.; Crispino, S.; Bouche, G.; Meheus, L.; Sukhatme, V.P. Repurposing drugs in oncology (ReDO)—Selective PDE5 inhibitors as anti-cancer agents. Ecancermedicalscience 2018, 12, 824. [Google Scholar] [CrossRef] [Green Version]
- Demoulin, B.; Hermant, M.; Castrogiovanni, C.; Staudt, C.; Dumont, P. Resveratrol induces DNA damage in colon cancer cells by poisoning topoisomerase II and activates the ATM kinase to trigger p53-dependent apoptosis. Toxicol. In Vitr. 2015, 29, 1156–1165. [Google Scholar] [CrossRef]
- Li, H.; Liu, L.; David, M.L.; Whitehead, C.M.; Chen, M.; Fetter, J.R.; Sperl, G.J.; Pamukcu, R.; Thompson, W.J. Pro-apoptotic actions of exisulind and CP461 in SW480 colon tumor cells involve β-catenin and cyclin D1 down-regulation. Biochem. Pharmacol. 2002, 64, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Mayur, Y.; Ahmad, O.; Rajendra Prasad, V.; Purohit, M.; Srinivasulu, N.; Shanta Kumar, S. Synthesis of 2-methyl N10-substituted acridones as selective inhibitors of multidrug resistance (MDR) associated protein in cancer cells. Med. Chem. 2008, 4, 457–465. [Google Scholar] [CrossRef]
- Ding, P.-R.; Tiwari, A.K.; Ohnuma, S.; Lee, J.W.K.K.; An, X.; Dai, C.-L.; Lu, Q.-S.; Singh, S.; Yang, D.-H.; Talele, T.T.; et al. The Phosphodiesterase-5 Inhibitor Vardenafil Is a Potent Inhibitor of ABCB1/P-Glycoprotein Transporter. PLoS ONE 2011, 6, e19329. [Google Scholar] [CrossRef]
- Shi, Z.; Tiwari, A.K.; Shukla, S.; Robey, R.W.; Singh, S.; Kim, I.-W.; Bates, S.E.; Peng, X.; Abraham, I.; Ambudkar, S.V. Sildenafil reverses ABCB1-and ABCG2-mediated chemotherapeutic drug resistance. Cancer Res. 2011, 71, 3029–3041. [Google Scholar] [CrossRef] [Green Version]
- Peak, T.C.; Richman, A.; Gur, S.; Yafi, F.A.; Hellstrom, W.J. The role of PDE5 inhibitors and the NO/cGMP pathway in cancer. Sex. Med. Rev. 2016, 4, 74–84. [Google Scholar] [CrossRef]
- Chang, J.-F.; Hsu, J.-L.; Sheng, Y.-H.; Leu, W.-J.; Yu, C.-C.; Chan, S.-H.; Chan, M.-L.; Hsu, L.-C.; Liu, S.-P.; Guh, J.-H. Phosphodiesterase type 5 (PDE5) inhibitors sensitize topoisomerase II inhibitors in killing prostate cancer through PDE5-independent impairment of HR and NHEJ DNA repair systems. Front. Oncol. 2019, 8, 681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [Green Version]
- Vos, S.M.; Tretter, E.M.; Schmidt, B.H.; Berger, J.M. All tangled up: How cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol. 2011, 12, 827–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailly, C. Contemporary challenges in the design of topoisomerase II inhibitors for cancer chemotherapy. Chem. Rev. 2012, 112, 3611–3640. [Google Scholar] [CrossRef]
- Macieja, A.; Kopa, P.; Galita, G.; Pastwa, E.; Majsterek, I.; Poplawski, T. Comparison of the effect of three different topoisomerase II inhibitors combined with cisplatin in human glioblastoma cells sensitized with double strand break repair inhibitors. Mol. Biol. Rep. 2019, 46, 3625–3636. [Google Scholar] [CrossRef] [Green Version]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, J.V.; Nitiss, J.L. DNA topoisomerase II as a target for cancer chemotherapy. Cancer Investig. 2002, 20, 570–589. [Google Scholar] [CrossRef] [PubMed]
- Skok, Z.i.; Zidar, N.; Kikelj, D.; Ilaš, J. Dual inhibitors of human DNA topoisomerase II and other cancer-related targets. J. Med. Chem. 2019, 63, 884–904. [Google Scholar] [CrossRef]
- Larsen, A.K.; Escargueil, A.E.; Skladanowski, A. Catalytic topoisomerase II inhibitors in cancer therapy. Pharmacol. Ther. 2003, 99, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, B.; Yang, T.; Wang, N.; Gao, C.; Tan, C.; Liu, H.; Jiang, Y. Synthesis and antiproliferative activity of 9-benzylamino-6-chloro-2-methoxy-acridine derivatives as potent DNA-binding ligands and topoisomerase II inhibitors. Eur. J. Med. Chem. 2016, 116, 59–70. [Google Scholar] [CrossRef]
- Brindisi, M.; Kessler, S.M.; Kumar, V.; Zwergel, C. Multi-target directed ligands for the treatment of cancer. Front. Oncol. 2022, 12, 980141. [Google Scholar] [CrossRef]
- Lang, X.; Li, L.; Chen, Y.; Sun, Q.; Wu, Q.; Liu, F.; Tan, C.; Liu, H.; Gao, C.; Jiang, Y. Novel synthetic acridine derivatives as potent DNA-binding and apoptosis-inducing antitumor agents. Bioorganic Med. Chem. 2013, 21, 4170–4177. [Google Scholar] [CrossRef]
- El-Sharkawy, L.Y.; El-Sakhawy, R.A.; Abdel-Halim, M.; Lee, K.; Piazza, G.A.; Ducho, C.; Hartmann, R.W.; Abadi, A.H. Design and synthesis of novel annulated thienopyrimidines as phosphodiesterase 5 (PDE5) inhibitors. Arch. Pharm. 2018, 351, 1800018. [Google Scholar] [CrossRef]
- Tinsley, H.N.; Gary, B.D.; Thaiparambil, J.; Li, N.; Lu, W.; Li, Y.; Maxuitenko, Y.Y.; Keeton, A.B.; Piazza, G.A. Colon Tumor Cell Growth–Inhibitory Activity of Sulindac Sulfide and Other Nonsteroidal Anti-Inflammatory Drugs Is Associated with Phosphodiesterase 5 InhibitionSulindac Inhibits PDE5. Cancer Prev. Res. 2010, 3, 1303–1313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Xi, Y.; Tinsley, H.N.; Gurpinar, E.; Gary, B.D.; Zhu, B.; Li, Y.; Chen, X.; Keeton, A.B.; Abadi, A.H. Sulindac Selectively Inhibits Colon Tumor Cell Growth by Activating the cGMP/PKG Pathway to Suppress Wnt/β-Catenin Signaling. Mol. Cancer Ther. 2013, 12, 1848–1859. [Google Scholar] [CrossRef] [Green Version]
- Shih, S.P.; Lee, M.G.; El-Shazly, M.; Juan, Y.S.; Wen, Z.H.; Du, Y.C.; Su, J.H.; Sung, P.J.; Chen, Y.C.; Yang, J.C.; et al. Tackling the Cytotoxic Effect of a Marine Polycyclic Quinone-Type Metabolite: Halenaquinone Induces Molt 4 Cells Apoptosis via Oxidative Stress Combined with the Inhibition of HDAC and Topoisomerase Activities. Mar. Drugs 2015, 13, 3132–3153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellón, R.F.; Mamposo, T.; Carrasco, R.; Rodés, L. Use of pyridine as cocatalyst for the synthesis of N-phenylanthranilic acids. Synth. Commun. 1996, 26, 3877–3883. [Google Scholar] [CrossRef]
- Nayak, S.; Panda, S.; Panda, P.; Padhy, S. Studies on acridone derivatives with and without inclusion complex formation with β-cyclodextrin. Bul. Chem. Comm. 2010, 42, 147–152. [Google Scholar]
- Boumendjel, A.; Macalou, S.; Ahmed-Belkacem, A.; Blanc, M.; Di Pietro, A. Acridone derivatives: Design, synthesis, and inhibition of breast cancer resistance protein ABCG2. Bioorganic Med. Chem. 2007, 15, 2892–2897. [Google Scholar] [CrossRef]
- Sabermahani, F.; Taher, M.A.; Bahrami, H. Separation and preconcentration of trace amounts of gold from water samples prior to determination by flame atomic absorption spectrometry. Arab. J. Chem. 2016, 9, S1700–S1705. [Google Scholar] [CrossRef] [Green Version]
- Sanfilippo, L.J. An improved synthesis of 9-CHLORO-2-METHOXYACRIDINE. Org. Prep. Proced. Int. 1991, 23, 130–132. [Google Scholar] [CrossRef]
- Kumar, R.; Sharma, A.; Sharma, S.; Silakari, O.; Singh, M.; Kaur, M. Synthesis, characterization and antitumor activity of 2-methyl-9-substituted acridines. Arab. J. Chem. 2017, 10, S956–S963. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.C.; Peng, B.R.; Hsu, K.C.; El-Shazly, M.; Shih, S.P.; Lin, T.E.; Kuo, F.W.; Chou, Y.C.; Lin, H.Y.; Lu, M.C. 13-Acetoxysarcocrassolide Exhibits Cytotoxic Activity Against Oral Cancer Cells Through the Interruption of the Keap1/Nrf2/p62/SQSTM1 Pathway: The Need to Move Beyond Classical Concepts. Mar. Drugs 2020, 18, 382. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Liu, Y.C.; El-Shazly, M.; Shih, S.P.; Du, Y.C.; Hsu, Y.M.; Lin, H.Y.; Chen, Y.C.; Wu, Y.C.; Yang, S.C.; et al. The Antioxidant from Ethanolic Extract of Rosa cymosa Fruits Activates Phosphatase and Tensin Homolog In Vitro and In Vivo: A New Insight on Its Antileukemic Effect. Int. J. Mol. Sci. 2019, 20, 1935. [Google Scholar] [CrossRef] [Green Version]
- ElHady, A.K.; Shih, S.-P.; Chen, Y.-C.; Liu, Y.-C.; Ahmed, N.S.; Keeton, A.B.; Piazza, G.A.; Engel, M.; Abadi, A.H.; Abdel-Halim, M. Extending the use of tadalafil scaffold: Development of novel selective phosphodiesterase 5 inhibitors and histone deacetylase inhibitors. Bioorganic Chem. 2020, 98, 103742. [Google Scholar] [CrossRef] [PubMed]
- Hafez, D.E.; Hafez, E.; Eddiasty, I.; Shih, S.-P.; Chien, L.-C.; Hong, Y.-J.; Lin, H.-Y.; Keeton, A.B.; Piazza, G.A.; Abdel-Halim, M. Novel thiazolidine derivatives as potent selective pro-apoptotic agents. Bioorganic Chem. 2021, 114, 105143. [Google Scholar] [CrossRef]
- Rabal, O.; Sánchez-Arias, J.A.; Cuadrado-Tejedor, M.; de Miguel, I.; Pérez-González, M.; García-Barroso, C.; Ugarte, A.; Estella-Hermoso de Mendoza, A.; Sáez, E.; Espelosin, M. Discovery of in vivo chemical probes for treating Alzheimer’s disease: Dual phosphodiesterase 5 (PDE5) and class I histone deacetylase selective inhibitors. ACS Chem. Neurosci. 2018, 10, 1765–1782. [Google Scholar] [CrossRef] [PubMed]
- Cuadrado-Tejedor, M.; Garcia-Barroso, C.; Sánchez-Arias, J.A.; Rabal, O.; Pérez-González, M.; Mederos, S.; Ugarte, A.; Franco, R.; Segura, V.; Perea, G. A first-in-class small-molecule that acts as a dual inhibitor of HDAC and PDE5 and that rescues hippocampal synaptic impairment in Alzheimer’s disease mice. Neuropsychopharmacology 2017, 42, 524–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, F.; Wang, H.; Ni, W.; Zheng, X.; Wang, M.; Bao, K.; Ling, D.; Li, X.; Xu, Y.; Zhang, H. Design, synthesis, and biological evaluation of orally available first-generation dual-target selective inhibitors of acetylcholinesterase (AChE) and phosphodiesterase 5 (PDE5) for the treatment of Alzheimer’s disease. ACS Chem. Neurosci. 2018, 9, 328–345. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Cpd. No. | R | Ar | PDE5 % Inhibition at 10 μM | PDE5 IC50 (μM) |
|---|---|---|---|---|
| 1 | -H | phenyl | 52.24 ± 5.87 | 9.33 ± 4.62 |
| 2 | -H | 2-tolyl | 50.35 ± 3.68 | 9.87 ± 2.06 |
| 3 | -H | 3-tolyl | 59.34 ± 6.18 | 6.40 ± 1.89 |
| 4 | -H | 4-tolyl | 73.98 ± 12.43 | 4.44 ± 1.82 |
| 5 | -H | 2-methoxyphenyl | 45.06 ± 4.54 | >10 |
| 6 | -H | 3-methoxyphenyl | 39.74 ± 3.20 | >10 |
| 7 | -H | 4-methoxyphenyl | NI | >10 |
| 8 | -H | 2-fluorophenyl | 57.53 ± 6.43 | 7.16 ± 2.75 |
| 9 | -H | 3-fluorophenyl | 55.94 ± 5.78 | 7.49 ± 2.20 |
| 10 | -H | 4-fluorophenyl | 46.97 ± 7.06 | >10 |
| 11 | -H | 2-chlorophenyl | 87.29 ± 3.68 | 4.33 ± 2.35 |
| 12 | -H | 3-chlorophenyl | 62.16 ± 4.94 | 6.29 ± 1.18 |
| 13 | -H | 4-chlorophenyl | 45.27 ± 3.99 | >10 |
| 14 | -H | 2-furanyl | 56.55 ± 7.38 | 7.43 ± 2.69 |
| 15 | -H | 2-thienyl | 58.43 ± 1.47 | 6.43 ± 1.30 |
| 16 | -H | 2,3-dimethoxyphenyl | 36.72 ± 8.14 | >10 |
| 17 | -H | 2,4-dimethoxyphenyl | 60.20 ± 5.89 | 6.77 ± 1.31 |
| 18 | -H | 3,4-dimethoxyphenyl | 51.16 ± 5.26 | 9.77 ± 3.11 |
| 19 | -H | 3-fluoro-4-methoxyphenyl | 79.24 ± 8.48 | 3.39 ± 1.03 |
| 20 | -H | 2,3-difluorphenyl | 57.86 ± 6.04 | 7.25 ± 2.21 |
| 21 | -H | 2,4-difluorphenyl | 31.93 ± 8.01 | >10 |
| 22 | -H | 3,4-difluorphenyl | 36.24 ± 9.78 | >10 |
| 23 | -H | phenylmethyl | 57.31 ± 3.58 | 6.68 ± 1.10 |
| 24 | -H | 4-(thiophen-2-yl)phenyl | 42.94 ± 5.35 | >10 |
| 25 | -OCH3 | phenyl | 77.23 ± 3.03 | 3.61 ± 1.00 |
| 26 | -OCH3 | 4-tolyl | 88.09 ± 4.28 | 0.83 ± 0.12 |
| 27 | -OCH3 | 4-methoxyphenyl | 88.77 ± 4.84 | 1.87 ± 1.03 |
| 28 | -OCH3 | 4-fluorophenyl | 64.25 ± 7.47 | 5.31 ± 1.27 |
| 29 | -OCH3 | 4-chlorophenyl | 66.07 ± 3.27 | 5.35 ± 1.01 |
| 30 | -CH3 | phenyl | 68.75 ± 7.05 | 4.52 ± 1.74 |
| 31 | -CH3 | 4-chlorophenyl | 65.67 ± 7.48 | 4.78 ± 1.48 |
| Cpd. No. | Mean % Growth Inhibition | Cpd. No. | Mean % Growth Inhibition |
|---|---|---|---|
| 1 | 62.74 | 17 | 60.80 |
| 2 | 77.65 | 18 | 81.58 |
| 3 | 77.21 | 19 | 76.16 |
| 4 | 74.79 | 20 | 40.73 |
| 5 | 60.77 | 21 | 56.35 |
| 6 | 63.07 | 22 | 64.84 |
| 7 | 63.25 | 23 | 60.71 |
| 8 | 56.47 | 24 | 96.88 |
| 9 | 64.19 | 25 | 80.05 |
| 10 | 72.34 | 26 | 79.9 |
| 11 | 78.36 | 27 | 87.94 |
| 12 | 72.98 | 28 | 92.16 |
| 13 | 59.43 | 29 | 93.27 |
| 14 | 38.41 | 30 | 64.5 |
| 16 | 63.87 | 31 | 66.88 |
| Cell Line | Colorectal Adenocarcinoma | Skin Fibroblast | ||
|---|---|---|---|---|
| HCT-116 | CCD966SK | |||
| Cpd. No. | % Inhibition at 10 µM | IC50 μM a | % Inhibition at 10 µM | IC50 μM a |
| 1 | 67.16 ± 2.40 | 2.81 ± 0.2 | 47.97 ± 1.96 | ND |
| 2 | 66.96 ± 0.14 | 4.01 ± 0.17 | 59.74 ± 0.55 | 4.18 ± 0.36 |
| 3 | 66.71 ± 2.10 | 4.60 ± 0.07 | 54.31 ± 0.98 | 8.51 ± 0.07 |
| 4 | 69.31 ± 2.09 | 3.59 ± 0.27 | 38.07 ± 5.90 | ND |
| 5 | 63.71 ± 0.62 | 6.83 ± 0.31 | 6.65 ± 1.18 | ND |
| 6 | 18.97 ± 4.43 | ND | 54.31 ± 0.98 | 8.51 ± 0.07 |
| 7 | 30.16 ± 9.09 | ND | 13.09 ± 2.52 | ND |
| 8 | 66.44 ± 0.80 | 3.05 ± 0.15 | 42.52 ± 2.0 | ND |
| 9 | 57.06 ± 1.36 | 7.30 ± 0.33 | 42.74 ± 2.09 | ND |
| 10 | 67.57 ± 0.29 | 2.75 ± 0.04 | 52.93 ± 1.00 | 9.26 ± 0.21 |
| 11 | 63.49 ± 1.20 | 7.13 ± 0.24 | 36.77 ± 3.49 | ND |
| 12 | 63.40 ± 7.54 | 4.84 ± 0.05 | 45.43 ± 0.42 | ND |
| 13 | 11.67 ± 4.59 | ND | 2.73 ± 4.11 | ND |
| 14 | 58.83 ± 1.72 | 8.41 ± 0.19 | 33.89 ± 0.85 | ND |
| 15 | 39.65 ± 4.72 | ND | 11.54 ± 0.04 | ND |
| 16 | 43.58 ± 2.65 | ND | 29.78 ± 2.20 | ND |
| 17 | 35.40 ± 3.74 | ND | 35.11 ± 0.42 | ND |
| 18 | 49.26 ± 0.88 | 9.74 ± 0.31 | 32.27 ± 0.58 | ND |
| 19 | 48.79 ± 1.04 | ND | 41.77 ± 0.99 | ND |
| 20 | 58.56 ± 3.21 | 4.96 ± 0.02 | 40.30 ± 3.56 | ND |
| 21 | 67.37 ± 0.67 | 3.95 ± 0.82 | 30.94 ± 0.86 | ND |
| 22 | 56.92 ± 0.61 | 6.99 ± 0.17 | 37.49 ± 7.38 | ND |
| 23 | 40.12 ± 0.11 | ND | 11.36 ± 7.38 | ND |
| 24 | 57.14 ± 3.10 | 7.00 ± 0.56 | 47.41 ± 2.48 | ND |
| 25 | 59.62 ± 2.66 | 7.43 ± 0.77 | 44.14 ± 3.80 | ND |
| 26 | 38.11 ± 1.35 | ND | 46.76 ± 3.21 | ND |
| 27 | 36.43 ± 7.55 | ND | 19.04 ± 3.94 | ND |
| 28 | 75.53 ± 2.52 | 2.51 ± 0.53 | 60.66 ± 2.63 | 4.67 ± 0.07 |
| 29 | 28.83 ± 2.64 | ND | 2.90 ± 2.51 | ND |
| 30 | 68.46 ± 1.55 | 5.78 ± 0.11 | 47.93 ± 1.06 | ND |
| 31 | 55.53 ± 1.69 | 7.95 ± 0.65 | 60.02 ± 3.07 | 4.96 ± 0.62 |
| cisplatin | 83.00 ± 0.38 | 3.62 ± 0.19 | 38.73 ± 5.19 | ND |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ammar, L.; Lin, H.-Y.; Shih, S.-P.; Tsai, T.-N.; Syu, Y.-T.; Abdel-Halim, M.; Hwang, T.-L.; Abadi, A.H. Novel 9-Benzylaminoacridine Derivatives as Dual Inhibitors of Phosphodiesterase 5 and Topoisomerase II for the Treatment of Colon Cancer. Molecules 2023, 28, 840. https://doi.org/10.3390/molecules28020840
Ammar L, Lin H-Y, Shih S-P, Tsai T-N, Syu Y-T, Abdel-Halim M, Hwang T-L, Abadi AH. Novel 9-Benzylaminoacridine Derivatives as Dual Inhibitors of Phosphodiesterase 5 and Topoisomerase II for the Treatment of Colon Cancer. Molecules. 2023; 28(2):840. https://doi.org/10.3390/molecules28020840
Chicago/Turabian StyleAmmar, Lina, Hung-Yu Lin, Shou-Ping Shih, Tsen-Ni Tsai, Yu-Ting Syu, Mohammad Abdel-Halim, Tsong-Long Hwang, and Ashraf H. Abadi. 2023. "Novel 9-Benzylaminoacridine Derivatives as Dual Inhibitors of Phosphodiesterase 5 and Topoisomerase II for the Treatment of Colon Cancer" Molecules 28, no. 2: 840. https://doi.org/10.3390/molecules28020840
APA StyleAmmar, L., Lin, H. -Y., Shih, S. -P., Tsai, T. -N., Syu, Y. -T., Abdel-Halim, M., Hwang, T. -L., & Abadi, A. H. (2023). Novel 9-Benzylaminoacridine Derivatives as Dual Inhibitors of Phosphodiesterase 5 and Topoisomerase II for the Treatment of Colon Cancer. Molecules, 28(2), 840. https://doi.org/10.3390/molecules28020840