

Novel 1,2,4-Triazole- and Tetrazole-Containing 4H-Thiopyrano[2,3-b]quinolines: Synthesis Based on the Thio-Michael/aza-Morita–Baylis–Hillman Tandem Reaction and Investigation of Antiviral Activity

 , , ,
, , ,  , , , , , and
, , , , , and
Abstract
:
1. Introduction
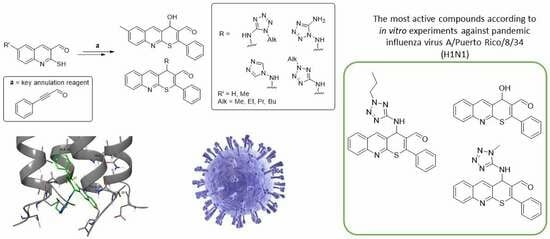
2. Results and Discussion
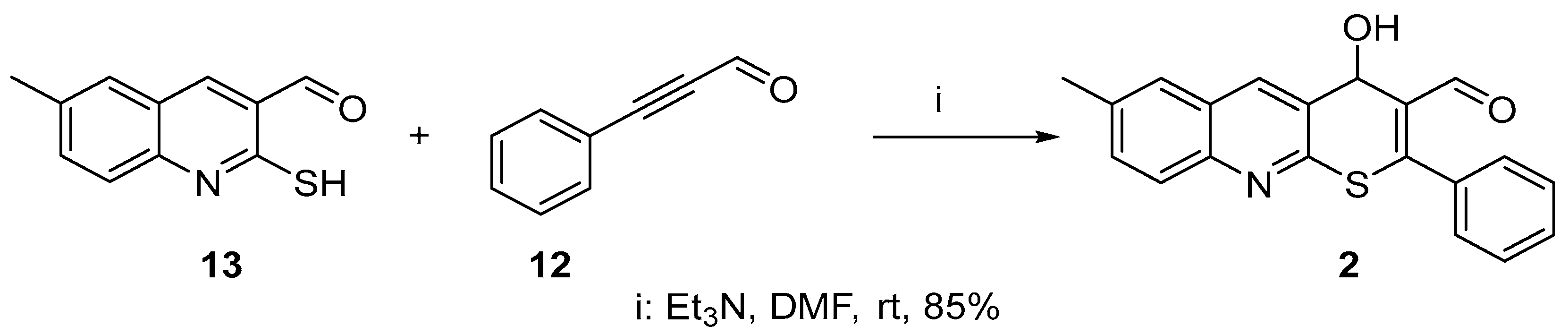
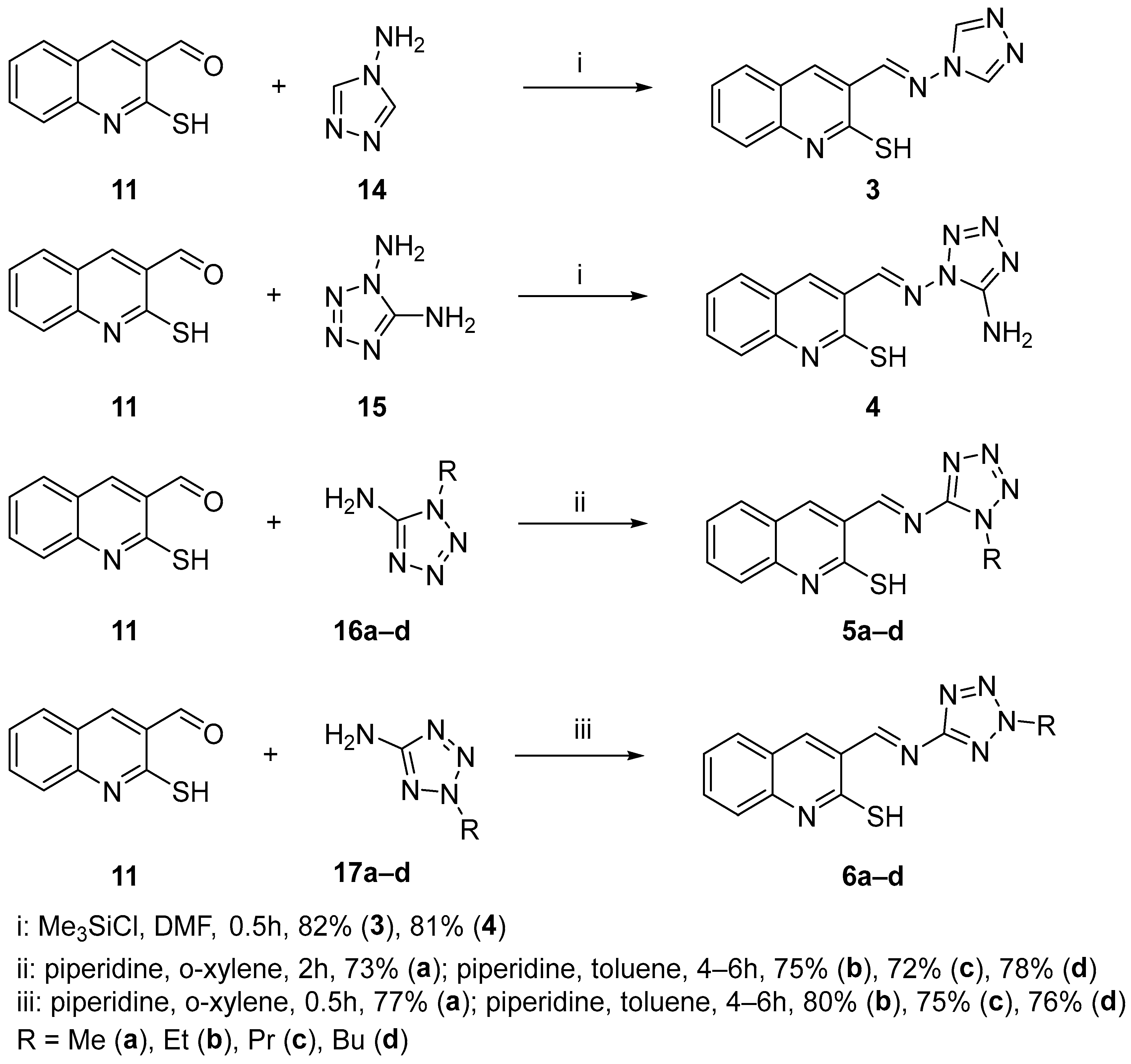
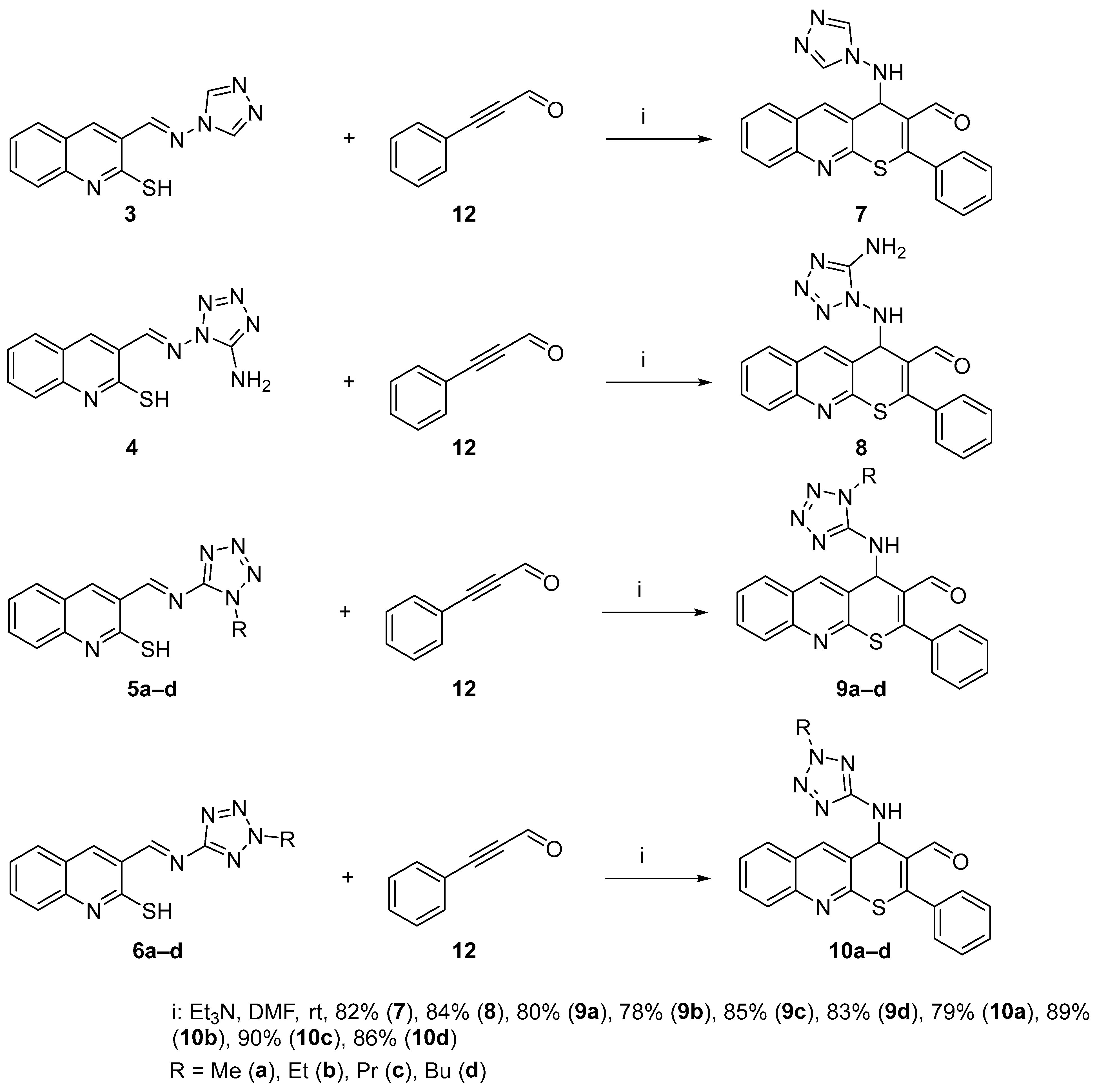
2.1. Chemistry
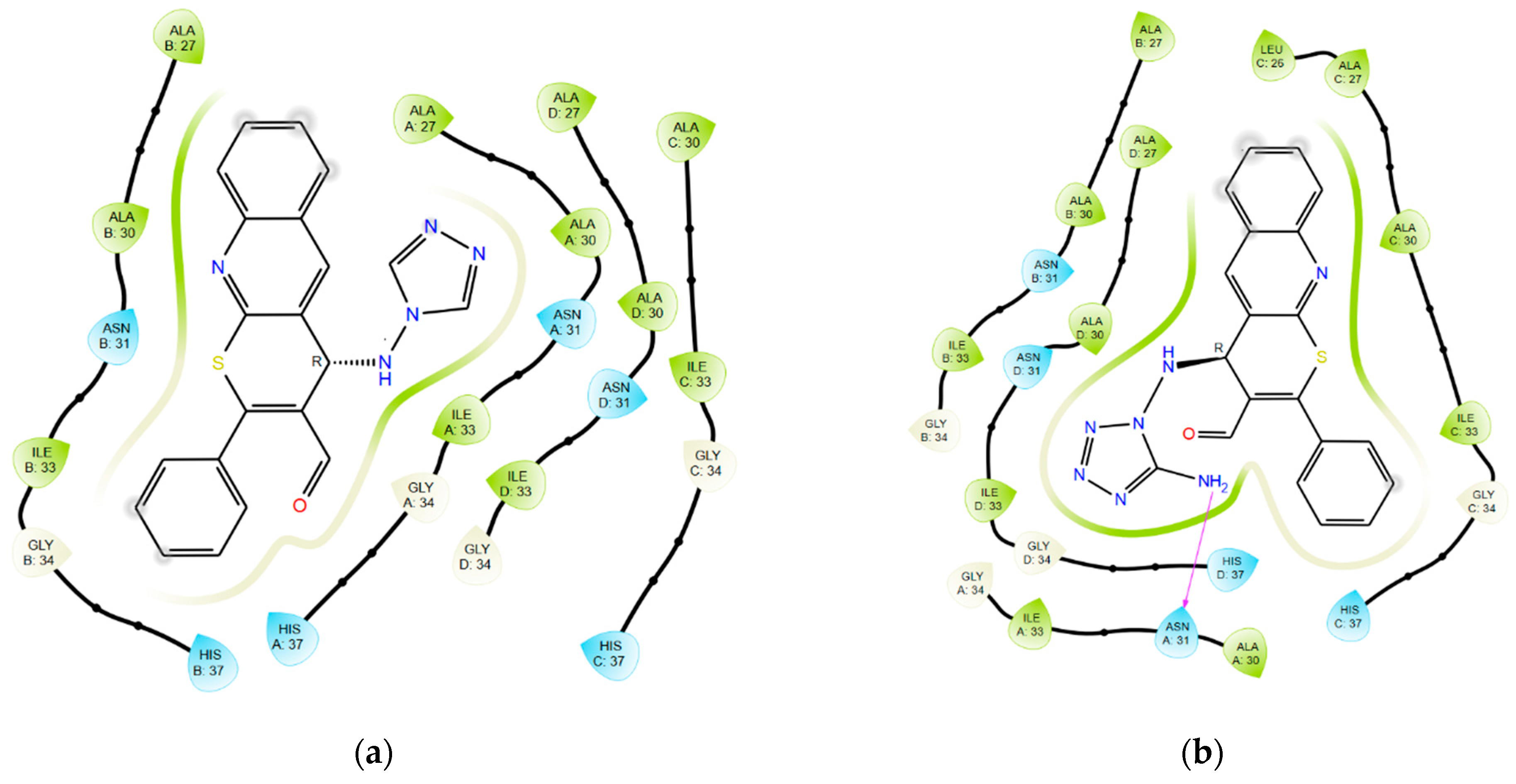
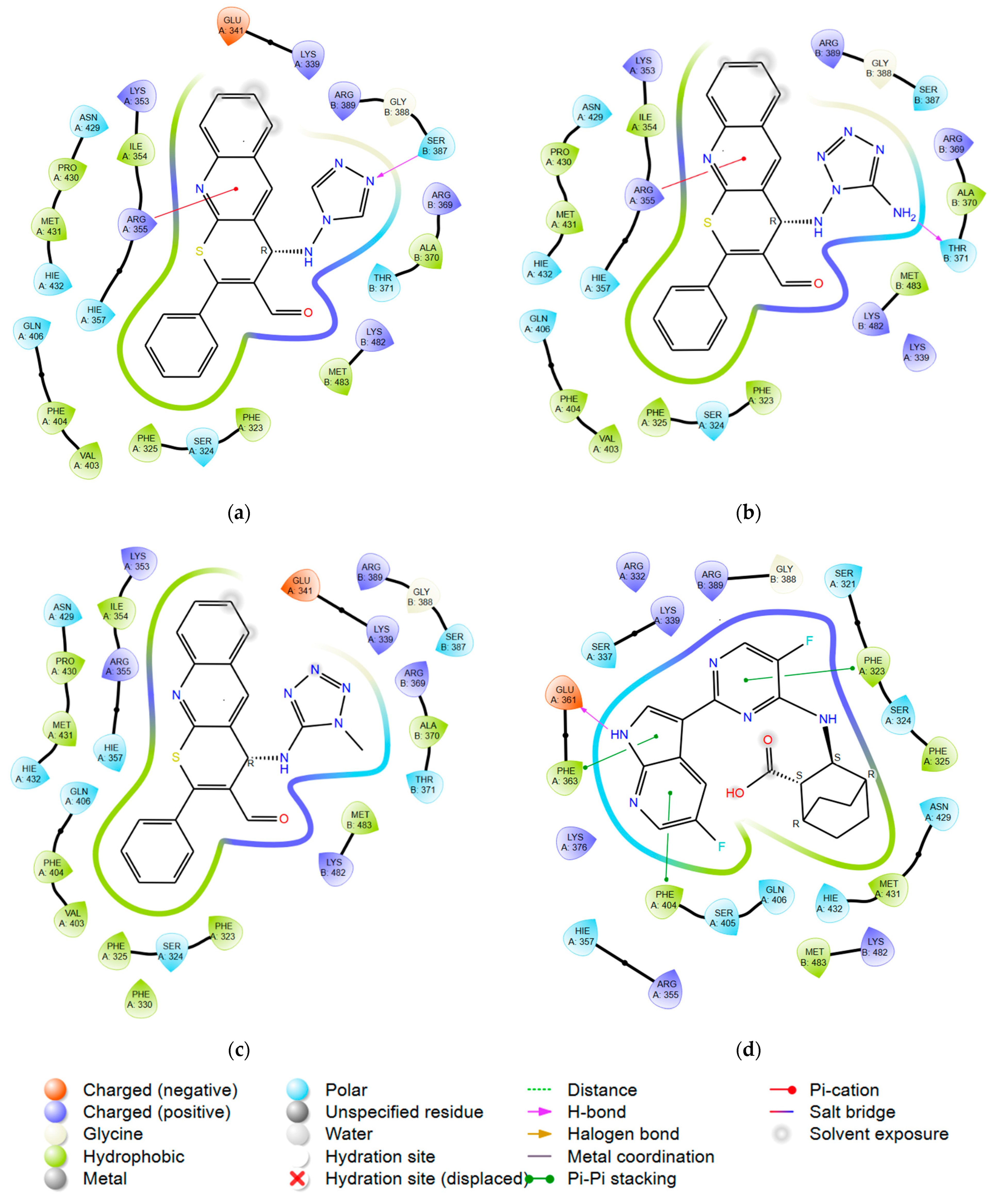
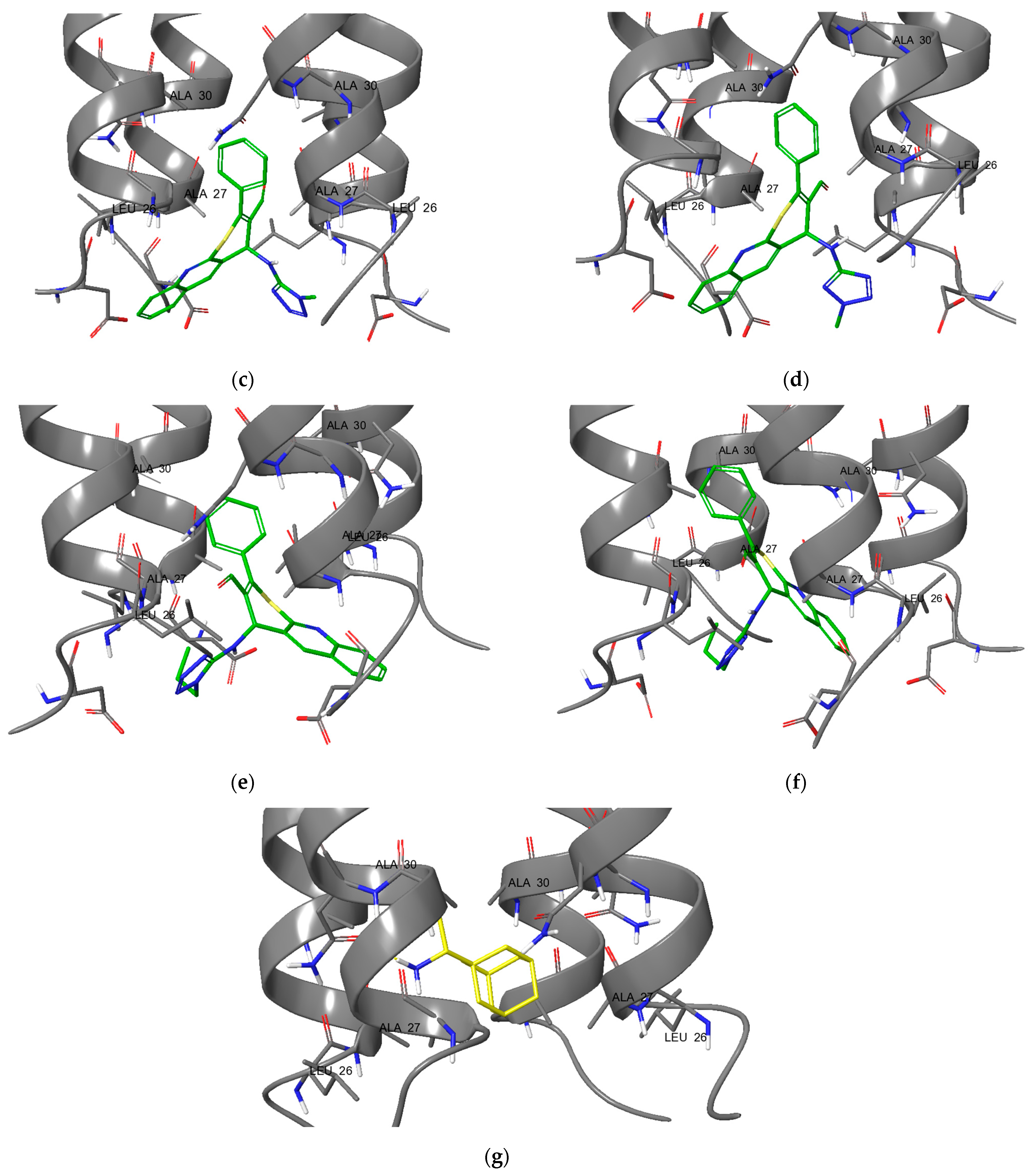
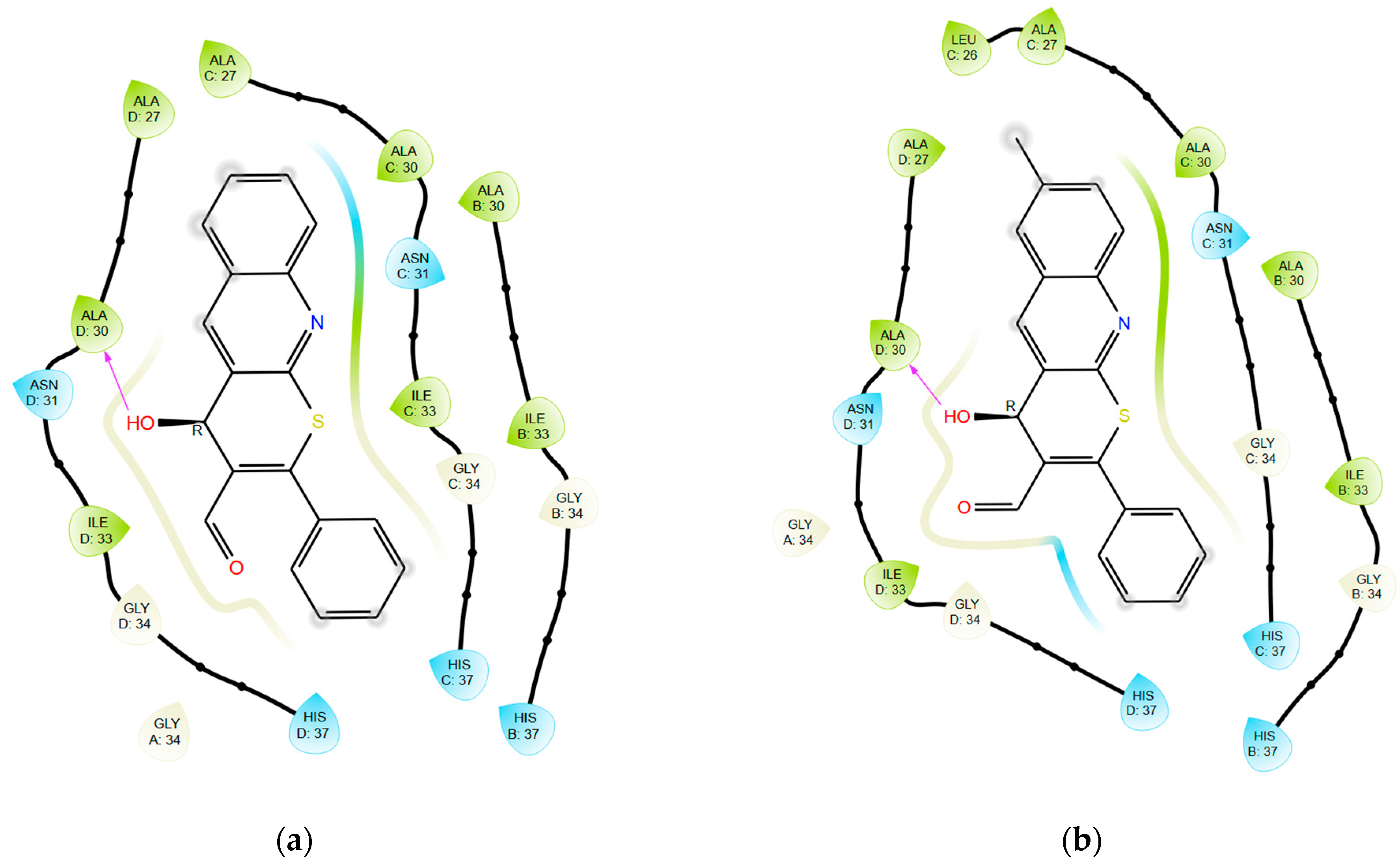
2.2. In Vitro Experiments and Target Validation
3. Materials and Methods
3.1. General Information
3.2. Chemistry
3.3. X-ray Diffraction
3.4. Biological Activity
3.4.1. Cytotoxicity Assay
3.4.2. CPE Reduction Assay
3.5. Molecular Modeling
3.5.1. Protein Structure Preparation
3.5.2. Ligand Structure Preparation
3.5.3. Molecular Docking: GridBox Building
3.5.4. Molecular Docking: Docking Procedure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bedford, J.; Farrar, J.; Ihekweazu, C.; Kang, G.; Koopmans, M.; Nkengasong, J. A New Twenty-First Century Science for Effective Epidemic Response. Nature 2019, 575, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Kiselev, O.I. Chemo Drugs and Influenza Chemotherapy; Rostok: St. Petersburg, Russia, 2012. [Google Scholar]
- Ostrovskii, V.A.; Popova, E.A.; Trifonov, R.E. Tetrazoles. In Comprehensive Heterocyclic Chemistry IV; Black, D.S., Cossy, J., Stevens, C.V., Eds.; Elsevier: Amsterdam, The Netherlands, 2022; Volume 6, pp. 182–232. [Google Scholar]
- Opsomer, T.; Dehaen, W. 1,2,4-Triazoles. In Comprehensive Heterocyclic Chemistry IV; Black, D.S., Cossy, J., Stevens, C.V., Eds.; Elsevier: Amsterdam, The Netherlands, 2022; Volume 5, pp. 78–121. [Google Scholar]
- McAteer, C.H.; Murugan, R.; Yamamoto, J.H. Pyridines and Their Benzo Derivatives: Applications. In Comprehensive Heterocyclic Chemistry IV; Black, D.S., Cossy, J., Stevens, C.V., Eds.; Elsevier: Amsterdam, The Netherlands, 2022; Volume 7, pp. 217–242. [Google Scholar]
- Popova, E.A.; Trifonov, R.E.; Ostrovskii, V.A. Tetrazoles for Biomedicine. Russ. Chem. Rev. 2019, 88, 644–676. [Google Scholar] [CrossRef]
- Pathania, S.; Narang, R.K.; Rawal, R.K. Role of Sulphur-Heterocycles in Medicinal Chemistry: An Update. Eur. J. Med. Chem. 2019, 180, 486–508. [Google Scholar] [CrossRef] [PubMed]
- Hou, L.; Zhang, Y.; Ju, H.; Cherukupalli, S.; Jia, R.; Zhang, J.; Huang, B.; Loregian, A.; Liu, X.; Zhan, P. Contemporary Medicinal Chemistry Strategies for the Discovery and Optimization of Influenza Inhibitors Targeting VRNP Constituent Proteins. Acta Pharm. Sin. B 2022, 12, 1805–1824. [Google Scholar] [CrossRef]
- De, A.; Sarkar, S.; Majee, A. Recent Advances on Heterocyclic Compounds with Antiviral Properties. Chem. Heterocycl. Compd. 2021, 57, 410–416. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Baloxavir Marboxil: A Review in Acute Uncomplicated Influenza. Drugs 2020, 80, 1109–1118. [Google Scholar] [CrossRef]
- Pécheur, E.-I.; Borisevich, V.; Halfmann, P.; Morrey, J.D.; Smee, D.F.; Prichard, M.; Mire, C.E.; Kawaoka, Y.; Geisbert, T.W.; Polyak, S.J. The Synthetic Antiviral Drug Arbidol Inhibits Globally Prevalent Pathogenic Viruses. J. Virol. 2016, 90, 3086–3092. [Google Scholar] [CrossRef]
- Sharma, S.; Singh, S. Molecular Docking Study for Binding Affinity of 2H-Thiopyrano[2,3-b]Quinoline Derivatives against CB1a. Interdiscip. Perspect. Infect. Dis. 2023, 2023, 1–10. [Google Scholar] [CrossRef]
- Nandeshwarappa, B.P.; Aruna Kumar, D.B.; Kumaraswamy, M.N.; Ravi Kumar, Y.S.; Bhojya Naik, H.S.; Mahadevan, K.M. Microwave Assisted Synthesis of Some Novel Thiopyrano[2,3-b]Quinolines as a New Class of Antimicrobial Agent. Phosphorus Sulfur Silicon Relat. Elem. 2006, 181, 1545–1556. [Google Scholar] [CrossRef]
- Kumar, S.V.; Muthusubramanian, S.; Perumal, S. Facile “on Water” Domino Reactions for the Expedient Synthesis of 2H-Thiopyrano[2,3-b]Quinolines. RSC Adv. 2015, 5, 30826–30832. [Google Scholar] [CrossRef]
- Sudol, M.; Fritz, J.L.; Tran, M.; Robertson, G.P.; Ealy, J.B.; Katzman, M. Evaluation of a System to Screen for Stimulators of Non-Specific DNA Nicking by HIV-1 Integrase: Application to a Library of 50,000 Compounds. Antivir. Chem. Chemother. 2011, 22, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Balkenhohl, F.; von dem Bussche-Hünnefeld, C.; Lansky, A.; Zechel, C. Combinatorial Synthesis of Small Organic Molecules. Angew. Chem. Int. Ed. 1996, 35, 2288–2337. [Google Scholar] [CrossRef]
- Knochel, P.; Molander, G.A. Comprehensive Organic Synthesis, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2014. [Google Scholar]
- Tietze, L.F. Domino Reactions: Concepts for Efficient Organic Synthesis; Walter de Gruyter GmbH: Berlin, Germany, 2014. [Google Scholar]
- Tietze, L.F. Domino Reactions in Organic Synthesis. Chem. Rev. 1996, 96, 115–136. [Google Scholar] [CrossRef] [PubMed]
- Basavaiah, D.; Venkateswara Rao, K.; Jannapu Reddy, R. The Baylis–Hillman Reaction: A Novel Source of Attraction, Opportunities, and Challenges in Synthetic Chemistry. Chem. Soc. Rev. 2007, 36, 1581–1588. [Google Scholar] [CrossRef]
- Alemán, J.; Núñez, A.; Marzo, L.; Marcos, V.; Alvarado, C.; García Ruano, J.L. Asymmetric Synthesis of 4-Amino-4H-Chromenes by Organocatalytic Oxa-Michael/Aza-Baylis-Hillman Tandem Reactions. Chem. Eur. J. 2010, 16, 9453–9456. [Google Scholar] [CrossRef]
- Khramchikhin, A.V.; Skryl’nikova, M.A.; Pavlyukova, Y.N.; Zarubaev, V.V.; Esaulkova, Y.L.; Muryleva, A.A.; Shmanyova, N.T.; Danagulyan, G.G.; Ostrovskii, V.A. Synthesis of Isomeric 4-(N-Methyltetrazolylamino)-2-Phenyl-4H-Thiopyrano[2,3-b]Quinoline-3-Carbaldehydes and 4-Hydroxy-2-Phenyl-4H-Thiopyrano[2,3-b]Quinoline-3-Carbaldehyde Based on Tandem Thiol-Michael and (Aza)-Morita–Baylis–Hillman Reactions and an in Vitro Study of the Activity of the Obtained Compounds against Influenza Virus. Chem. Heterocycl. Compd. 2022, 58, 267–270. [Google Scholar] [CrossRef]
- Zhong, N.-J.; Wang, Y.-Z.; Cheng, L.; Wang, D.; Liu, L. Recent Advances in the Annulation of Morita–Baylis–Hillman Adducts. Org. Biomol. Chem. 2018, 16, 5214–5227. [Google Scholar] [CrossRef] [PubMed]
- Chandra Bharadwaj, K. Intramolecular Morita–Baylis–Hillman and Rauhut–Currier Reactions. A Catalytic and Atom Economic Route for Carbocycles and Heterocycles. RSC Adv. 2015, 5, 75923–75946. [Google Scholar] [CrossRef]
- Ryabukhin, S.V.; Panov, D.M.; Plaskon, A.S.; Chuprina, A.; Pipko, S.E.; Tolmachev, A.A.; Shivanyuk, A.N. Combinatorial Synthesis of Chemical Building Blocks 1. Azomethines. Mol. Divers. 2012, 16, 625–637. [Google Scholar] [CrossRef]
- Henry, R.A.; Finnegan, W.G. Mono-Alkylation of Sodium 5-Aminotetrazole in Aqueous Medium. J. Am. Chem. Soc. 1954, 76, 923–926. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, Q.; Wang, M.-X. Multicomponent Reactions in Organic Synthesis; Wiley: New York, NY, USA, 2014. [Google Scholar]
- Poroikov, V.V. Computer-Aided Drug Design: From Discovery of Novel Pharmaceutical Agents to Systems Pharmacology. Biomeditsinskaya Khimiya 2020, 66, 30–41. [Google Scholar] [CrossRef] [PubMed]
- Filimonov, D.A.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.V.; Druzhilovskii, D.S.; Pogodin, P.V.; Poroikov, V.V. Prediction of the Biological Activity Spectra of Organic Compounds Using the Pass Online Web Resource. Chem. Heterocycl. Compd. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Lagunin, A.; Stepanchikova, A.; Filimonov, D.; Poroikov, V. PASS: Prediction of Activity Spectra for Biologically Active Substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef]
- Gamblin, S.J.; Haire, L.F.; Russell, R.J.; Stevens, D.J.; Xiao, B.; Ha, Y.; Vasisht, N.; Steinhauer, D.A.; Daniels, R.S.; Elliot, A.; et al. The Structure and Receptor Binding Properties of the 1918 Influenza Hemagglutinin. Science 2004, 303, 1838–1842. [Google Scholar] [CrossRef] [PubMed]
- Hanke, L.; Knockenhauer, K.E.; Brewer, R.C.; van Diest, E.; Schmidt, F.I.; Schwartz, T.U.; Ploegh, H.L. The Antiviral Mechanism of an Influenza A Virus Nucleoprotein-Specific Single-Domain Antibody Fragment. mBio 2016, 7, e01569-16. [Google Scholar] [CrossRef] [PubMed]
- Fudo, S.; Yamamoto, N.; Nukaga, M.; Odagiri, T.; Tashiro, M.; Hoshino, T. Two Distinctive Binding Modes of Endonuclease Inhibitors to the N-Terminal Region of Influenza Virus Polymerase Acidic Subunit. Biochemistry 2016, 55, 2646–2660. [Google Scholar] [CrossRef]
- Borisevich, S.S.; Gureev, M.A.; Yarovaya, O.I.; Zarubaev, V.V.; Kostin, G.A.; Porozov, Y.B.; Salakhutdinov, N.F. Can Molecular Dynamics Explain Decreased Pathogenicity in Mutant Camphecene-Resistant Influenza Virus? J. Biomol. Struct. Dyn. 2022, 40, 5481–5492. [Google Scholar] [CrossRef]
- Zhao, L.; Che, J.; Zhang, Q.; Li, Y.; Guo, X.; Chen, L.; Li, H.; Cao, R.; Li, X. Identification of Novel Influenza Polymerase PB2 Inhibitors Using a Cascade Docking Virtual Screening Approach. Molecules 2020, 25, 5291. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and Ligand Preparation: Parameters, Protocols, and Influence on Virtual Screening Enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Schnell, J.R.; Chou, J.J. Structure and Mechanism of the M2 Proton Channel of Influenza A Virus. Nature 2008, 451, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Bas, D.C.; Rogers, D.M.; Jensen, J.H. Very Fast Prediction and Rationalization of PKa Values for Protein-Ligand Complexes. Proteins Struct. Funct. Genet. 2008, 73, 765–783. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef] [PubMed]
- Repasky, M.P.; Shelley, M.; Friesner, R.A. Flexible Ligand Docking with Glide. Curr. Protoc. Bioinform. 2007, 18, 8–12. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | N | a CC50, µM | b IC50, µM | c SI | Compound | N | a CC50, µM | b IC50, µM | c SI |
|---|---|---|---|---|---|---|---|---|---|
 | 1 | >900 | 30 ± 5 | 31 |  | 9c | >700 | 293.8 ± 30.8 | >2 |
 | 2 | >900 | 293.9 ± 31.3 | >3 |  | 9d | >650 | 384.1 ± 42.1 | >2 |
 | 7 | >750 | 161.4 ± 10.9 | >5 |  | 10a | 549 ± 26.6 | >249.7 | <2 |
 | 8 | >750 | 190.3 ± 26.4 | >4 |  | 10b | >700 | 605.6 ± 62.9 | >1 |
 | 9a | >750 | 46 ± 5 | >16 |  | 10c | >700 | 18.4 ± 2.7 | >38 |
 | 9b | >700 | >700 | 1 |  | 10d | 68.9 ± 42.4 | >24.9 | <3 |
| Rimantadine | 312.3 ± 22.8 | 64.1 ± 7.2 | 5 | Oseltamivircarboxylate | >100 | 0.17 ± 0.02 | >588 |
| Compound | GlideScore@Target (kcal/mol) | Activity (IC50) | |||||
|---|---|---|---|---|---|---|---|
| NA | M2 | HA | NP | PB2 | PA | ||
| 1 | −4.61 | −7.90 | - | −4.26 | −5.33 | −4.87 | 30 ± 5 |
| 2 | −4.25 | −5.90 | - | −4.21 | −5.30 | −5.10 | 293.9 ± 31.3 |
| 7 | −3.99 | −7.79 | - | −3.71 | −6.41 | −4.71 | 161.4 ± 10.9 |
| 8 | −4.45 | −7.95 | - | −3.65 | −6.23 | −4.84 | 190.3 ± 26.4 |
| 9a | −4.34 | −8.34 | - | −3.16 | −6.37 | −4.62 | 46 ± 5 |
| 9b | −3.74 | −4.05 | - | −3.80 | −4.42 | −4.73 | >700 |
| 9c | −4.06 | −3.88 | - | −2.75 | −5.11 | −5.15 | 293.8 ± 30.8 |
| 9d | −3.87 | −3.51 | - | −2.32 | - | −5.15 | 384.1 ± 42.1 |
| 10a | −3.95 | −3.99 | - | −3.28 | −4.26 | −5.07 | >249.7 |
| 10b | −3.62 | −5.98 | - | −2.95 | −5.27 | −4.30 | 605.6 ± 62.9 |
| 10c | −3.40 | −5.08 | - | −3.92 | −5.33 | −4.57 | 18.4 ± 2.7 |
| 10d | −3.26 | −4.90 | - | −3.37 | −5.00 | −4.57 | >24.9 |
| Control | −5.50 | −6.60 | −7.10 | −6.56 | −6.05 | −6.56 | - |
| Compound | Glidescore, (kcal/mol) | MMGBSA ΔG, (kcal/mol) | IC50 | ||
|---|---|---|---|---|---|
| Neuraminidase | M2 | M2—Lipo | M2—Strain Energy | ||
| 1 | −4.61 | −7.90 | −14.07 | 0.96 | 30 ± 5 |
| 2 | −4.25 | −5.90 | −15.04 | 2.08 | 293.9 ± 31.3 |
| 9a | −4.34 | −8.34 | −11.42 | 2.02 | 46 ± 5 |
| 10a | −3.74 | −3.99 | −12.81 | 2.58 | >249.7 |
| Rimantadine | - | −6.60 | −13.25 | 0.78 | 64.1 ± 7.2 |
| Oseltamivir | −5.50 | - | - | - | 0.17 ± 0.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khramchikhin, A.V.; Skryl’nikova, M.A.; Gureev, M.A.; Zarubaev, V.V.; Esaulkova, I.L.; Ilyina, P.A.; Mammeri, O.A.; Spiridonova, D.V.; Porozov, Y.B.; Ostrovskii, V.A. Novel 1,2,4-Triazole- and Tetrazole-Containing 4H-Thiopyrano[2,3-b]quinolines: Synthesis Based on the Thio-Michael/aza-Morita–Baylis–Hillman Tandem Reaction and Investigation of Antiviral Activity. Molecules 2023, 28, 7427. https://doi.org/10.3390/molecules28217427
Khramchikhin AV, Skryl’nikova MA, Gureev MA, Zarubaev VV, Esaulkova IL, Ilyina PA, Mammeri OA, Spiridonova DV, Porozov YB, Ostrovskii VA. Novel 1,2,4-Triazole- and Tetrazole-Containing 4H-Thiopyrano[2,3-b]quinolines: Synthesis Based on the Thio-Michael/aza-Morita–Baylis–Hillman Tandem Reaction and Investigation of Antiviral Activity. Molecules. 2023; 28(21):7427. https://doi.org/10.3390/molecules28217427
Chicago/Turabian StyleKhramchikhin, Andrey V., Mariya A. Skryl’nikova, Maxim A. Gureev, Vladimir V. Zarubaev, Iana L. Esaulkova, Polina A. Ilyina, Oussama Abdelhamid Mammeri, Dar’ya V. Spiridonova, Yuri B. Porozov, and Vladimir A. Ostrovskii. 2023. "Novel 1,2,4-Triazole- and Tetrazole-Containing 4H-Thiopyrano[2,3-b]quinolines: Synthesis Based on the Thio-Michael/aza-Morita–Baylis–Hillman Tandem Reaction and Investigation of Antiviral Activity" Molecules 28, no. 21: 7427. https://doi.org/10.3390/molecules28217427
APA StyleKhramchikhin, A. V., Skryl’nikova, M. A., Gureev, M. A., Zarubaev, V. V., Esaulkova, I. L., Ilyina, P. A., Mammeri, O. A., Spiridonova, D. V., Porozov, Y. B., & Ostrovskii, V. A. (2023). Novel 1,2,4-Triazole- and Tetrazole-Containing 4H-Thiopyrano[2,3-b]quinolines: Synthesis Based on the Thio-Michael/aza-Morita–Baylis–Hillman Tandem Reaction and Investigation of Antiviral Activity. Molecules, 28(21), 7427. https://doi.org/10.3390/molecules28217427