2.1. Plant Triterpenoids of the Lupane Family
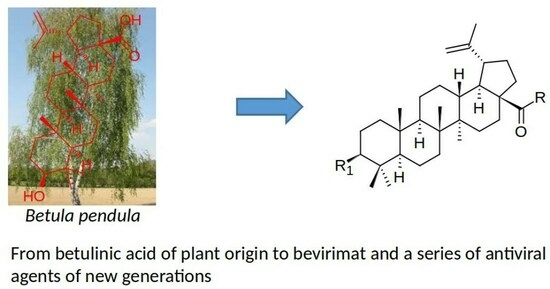
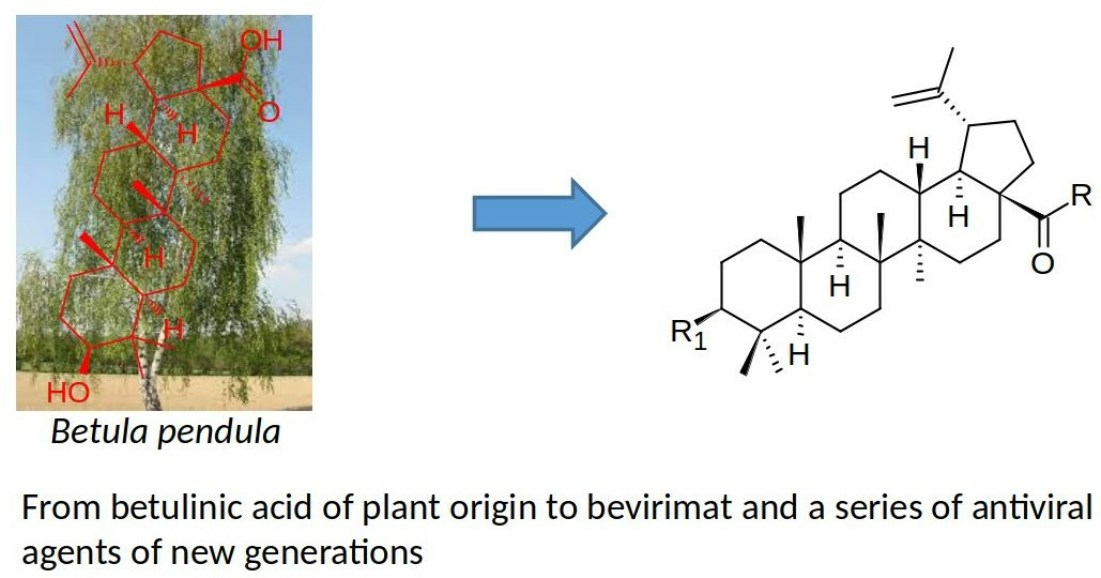
Betulin (
1;
Figure 1), a pentacyclic natural triterpenoid, represents one of the potent and for a long time known plant-derived products [
28]. Mostly, it has been found in the bark of various birch (
Betula) species and can be extracted therefrom. The biological effects of betulin (
1) have been intensively investigated, and have resulted in discovering its wide-ranging biological activity that involves antiviral, antibacterial, anticancer, and anti-inflammatory effects [
29]. The antiviral properties of betulin (
1) and its derivatives have been explored in the context of many different viruses [
30,
31,
32]. Betulinic acid (
2;
Figure 1) represents another plant product extractable from the bark of birch and from other plant sources [
33]. It can also be prepared synthetically by oxidation of betulin (
1) [
34]. The pharmacological characteristics of betulinic acid (
2) are similar to betulin (
1) [
35,
36,
37]. Bevirimat, 3-
O-(3′,3′-dimethylsuccinyl)betulinic acid (
3;
Figure 1), was one of the most promising derivatives of betulinic acid (
2). It has displayed potent anti-HIV activity with a novel mechanism of action [
38]. Unfortunately, its further development was terminated at Phase IIb of clinical trials due to the reduced efficacy of the compound against certain HIV strains [
39]. Nevertheless, the discovery of bevirimat (
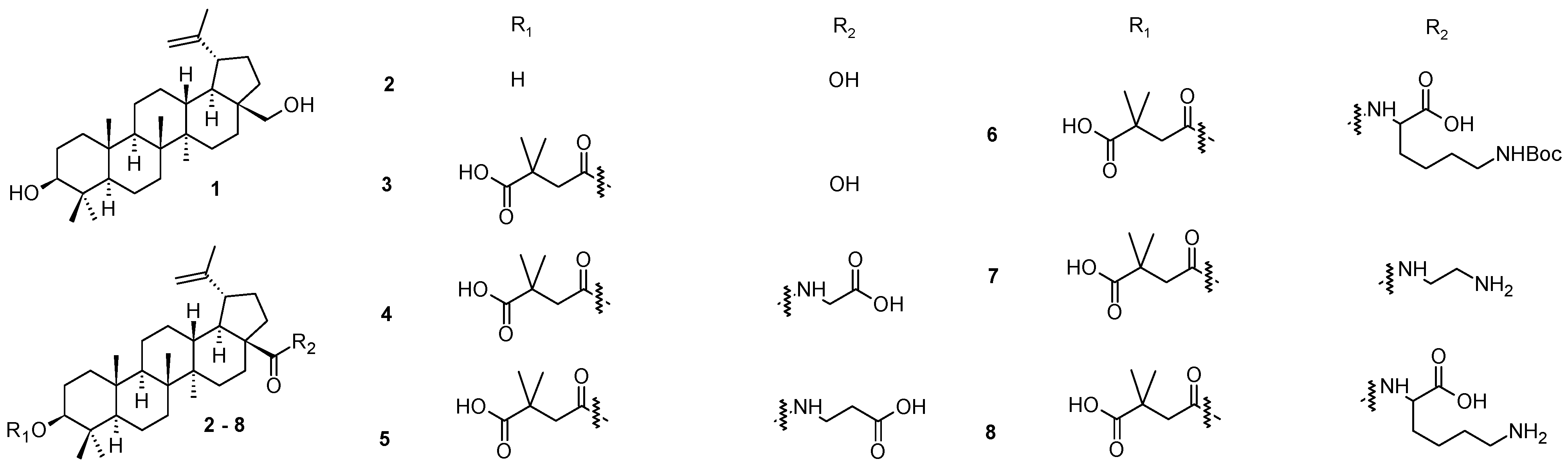
3) initiated a subsequent investigation in the field of plant triterpenoids and their derivatives bearing various functional groups or structural modifiers, resulting in a synthesis of several early but successful bevirimat analogs (
4–
8;
Figure 1) [
40,
41,
42]. However, currently, approved antiviral drugs have very diverse structures [
28,
43].
Bevirimat (
3) was the first-in-class HIV-1 maturation inhibitor. It showed a low efficacy, essentially due to the natural polymorphism of its target, the CA-SP1 junction. Moreover, its low solubility in water and in the physiological environment makes it difficult to study its interaction with the CA-SP1 junction. Therefore, designing new derivatives of bevirimat (
3) was performed by introducing different hydrophilic substituents at the C-28 carboxyl group to improve the solubility of the novel compounds in aqueous media. A synthesis of the novel derivatives, the effect of substituents at the C-28 carboxyl group, and their solubility in aqueous media were investigated intensively, and the ability of these molecules to inhibit viral infection and their cytotoxicity was carefully evaluated [
43]. Compared to the well-known bevirimat (
3), one of the prepared compounds (
7) showed higher solubility in aqueous media associated with a 2.5-fold increase in activity, higher selectivity index, and a better antiviral profile (
Table 1) [
43]. Moreover, for the first time, a direct interaction between the prepared compound (
7) and the domain CA-SP1 was shown by the NMR (nuclear magnetic resonance) study [
43]. Bevirimat (
3) was launched by several pharmaceutical companies for further development and commercialization [
44,
45]. However, while bevirimat (
3) succeeded in Phase IIa of the clinical trials, results obtained in Phase IIb of the clinical trials stopped the development of this new class of anti-HIV drug, principally because of the natural polymorphism of the CA-SP1 junction that led to a natural resistance of the virus to maturation inhibitors. The prepared pioneer bevirimat-based compounds (
4–
8;
Figure 1) represented novel, attractive, and promising agents for the future development of the next generation of HIV-1 maturation inhibitors [
43]. Their anti-HIV-1 effects are summarized in
Table 1.
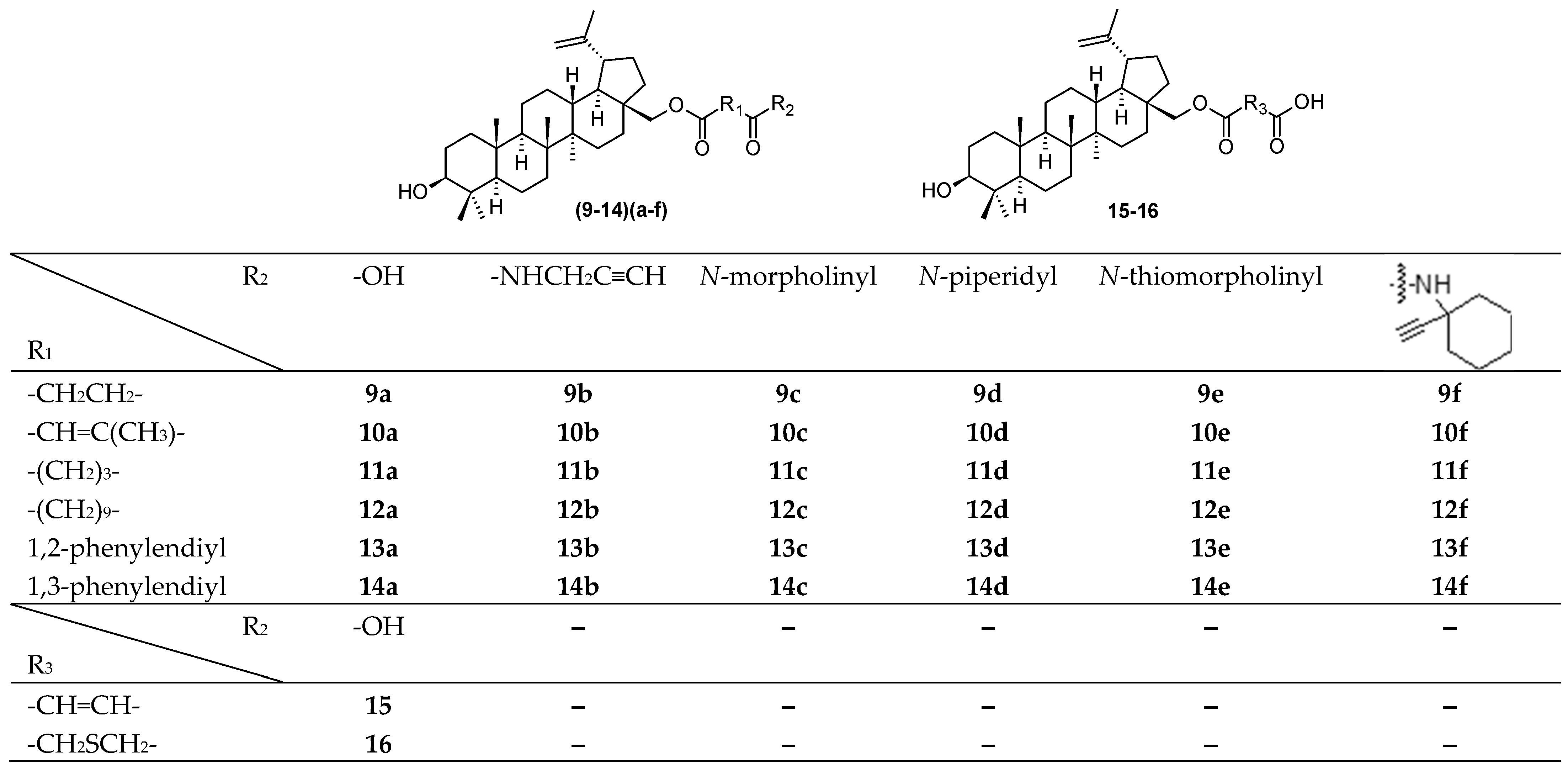
A search for new methods of antiviral therapy has been primarily focused on the use of substances of natural origin. Recently, a synthesis of a novel series of betulinic acid ester derivatives (
9–
14)(
a–
f) and
15–
16 (
Figure 2) was published [
28]. The structures of the novel compounds were established, and the compounds were tested against DNA and RNA viruses, for antiviral activity against several types of viruses, including HSV-1 [
28]. Antiviral experiments and monitoring of the time of addition of the active compound confirmed a research hypothesis and showed high antiviral effect of several derivatives against BEV, H1N1, and HSV-1. Compound
10d exhibited 6-fold more potent activity against HSV-1 (EC
50 = 17.2 µM) than the reference drug (acyclovir; EC
50 = 111.1 µM) (
Table 2). Compound
13e possessed the highest selectivity index (SI = 11.8) even when compared with that of acyclovir (SI = 14.0). Overall, all active compounds showed high virus-specific activity, as none of them showed activity in more than one virus. Most of the active compounds were active at the later steps of the replication cycle. This finding resulted in a suggestion of a mode of action during the step of nucleic acids/protein synthesis, assembly, or maturation. The in silico study was in good agreement with the in vitro data, confirming a high affinity of
10d to HSV-1 DNA polymerase. In addition, all ester and amide derivatives were tested for antiproliferative activity in A549 and MDCK (Madin-Darby canine kidney) cell lines (
Table 3). Ester derivatives in the series of (
9–
14)(
a–
f) and
15–
16, glutaric acid amides
11c and
11d, and succinic acid amides
9b and
9c showed high cytotoxicity values. These findings provided valuable data for further investigation of the active compounds, and for a subsequent betulin derivation to design novel compounds having higher potential to display antiviral activity. The results indicated that natural resources have still been one of the most important sources of priority structures in a search for new drug candidates.
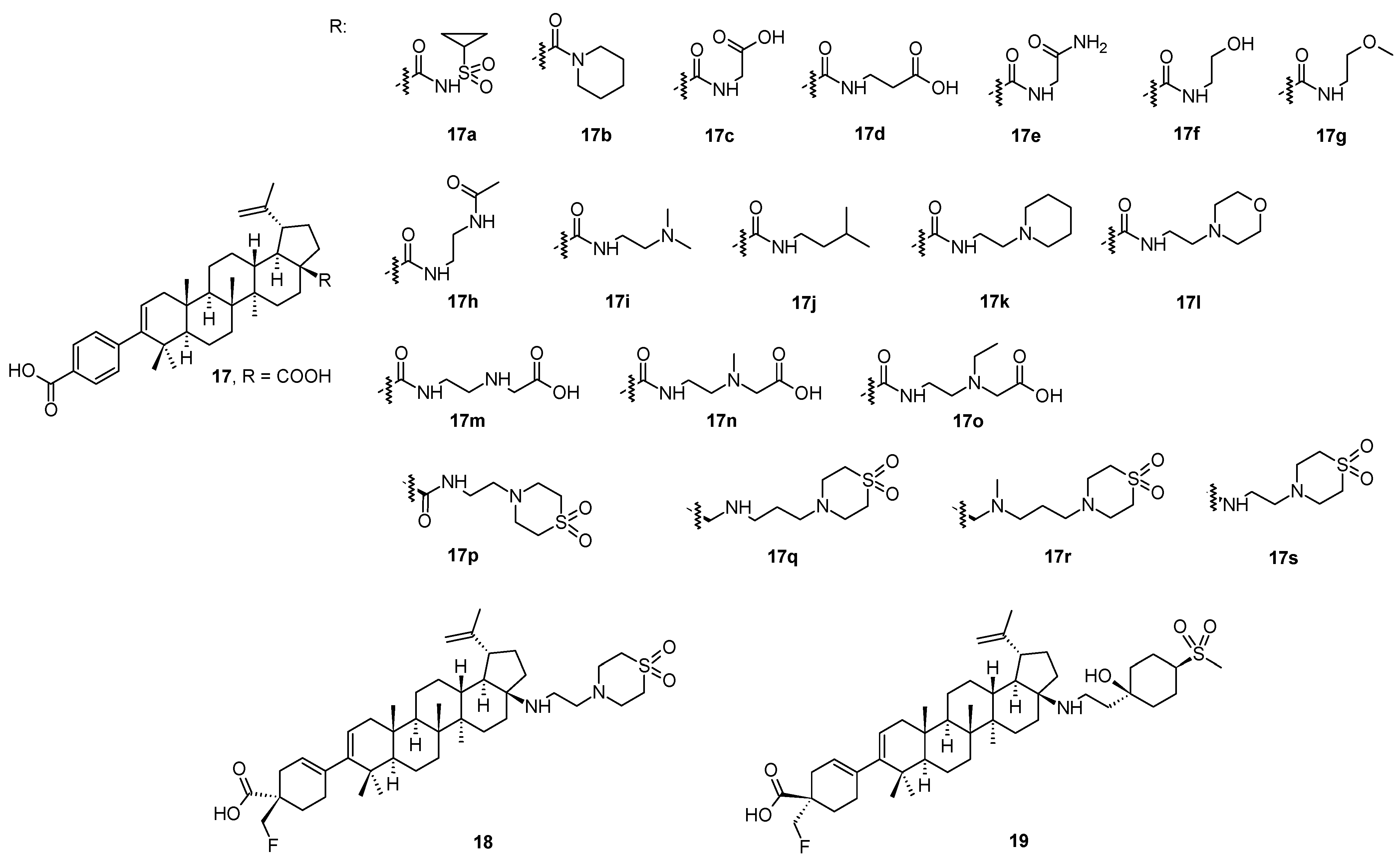
In a continuing search for novel HIV-1 maturation inhibitors, another series of promising compounds were designed and synthesized on the basis of a number of triterpenoid derivatives (
17–
17s;
Figure 3), namely the compound
18 (also known GSK3640254 or fipravirimat;
Figure 3) [
46]. Compound
18 exhibited significantly improved antiviral activity toward a range of clinically relevant polymorphic variants with reduced sensitivity toward the second-generation maturation inhibitor (
17s; also known as GSK3532795 or BMS-955176). The key structural difference between
18 and its earlier developed analogs (
17–
17s) is the replacement of the
para-substituted benzoic acid moiety located at the C-3 position of the triterpenoid skeleton with a cyclohex-3-ene-1-carboxylic acid substituted with a CH
2F moiety with the given absolute configuration (
18;
Figure 3). The sp
3 carbon atom at this site of the molecule provided a new vector for structure-activity relationship exploration and resulted in the identification of compounds with improved polymorphic characteristics while preserving the pharmacokinetic properties of the prototype. This structural element provided a new vector for exploring structure-activity relationships. The approach to the design of
18, the development of a synthetic route, and its preclinical profile were clearly described in detail in the original paper [
46]. Compound
18 has completed the Phase IIa of the clinical trials, in which it demonstrated a dose-related reduction in plasma HIV-1 RNA over 7–10 days, and the compound has been advanced into Phase IIb studies [
46].
The investigation of the structure-activity relationships of a series of HIV-1 maturation inhibitors based on the compound
17s (
Figure 3) continued by the subsequent incorporation of novel C-17 amine substituents to reduce the overall basicity of the target compounds [
47]. A replacement of the amine group on the C-17 side chain present in
17s with a tertiary alcohol in combination with either a heterocyclic ring system or a cyclohexyl ring substituted with polar groups provided potent wild-type (WT) HIV-1 maturation inhibitors. They also preserved excellent potency against a T332S/V362I/prR41G variant, a laboratory strain that served as a substitute to assess HIV-1 polymorphic virus characteristics [
47]. Compound
19 exhibited a broad anti-HIV-1 activity against relevant Gag polymorphic viruses, and displayed the most desirable overall profile in this series of the studied compounds. In pharmacokinetic studies,
19 had low acquittal and exhibited sufficient oral bioavailability in rats and dogs.
Compounds
18 and
19 had the most desirable overall profile in this series and were evaluated in rat and dog pharmacokinetic studies [
47,
48]. A comparison of basic anti-HIV-1 activity values is shown in
Table 4. An overall summary of antiviral activity values of compounds
17–
17s,
18, and
19 is presented in
Table 5 [
46,
47,
48].
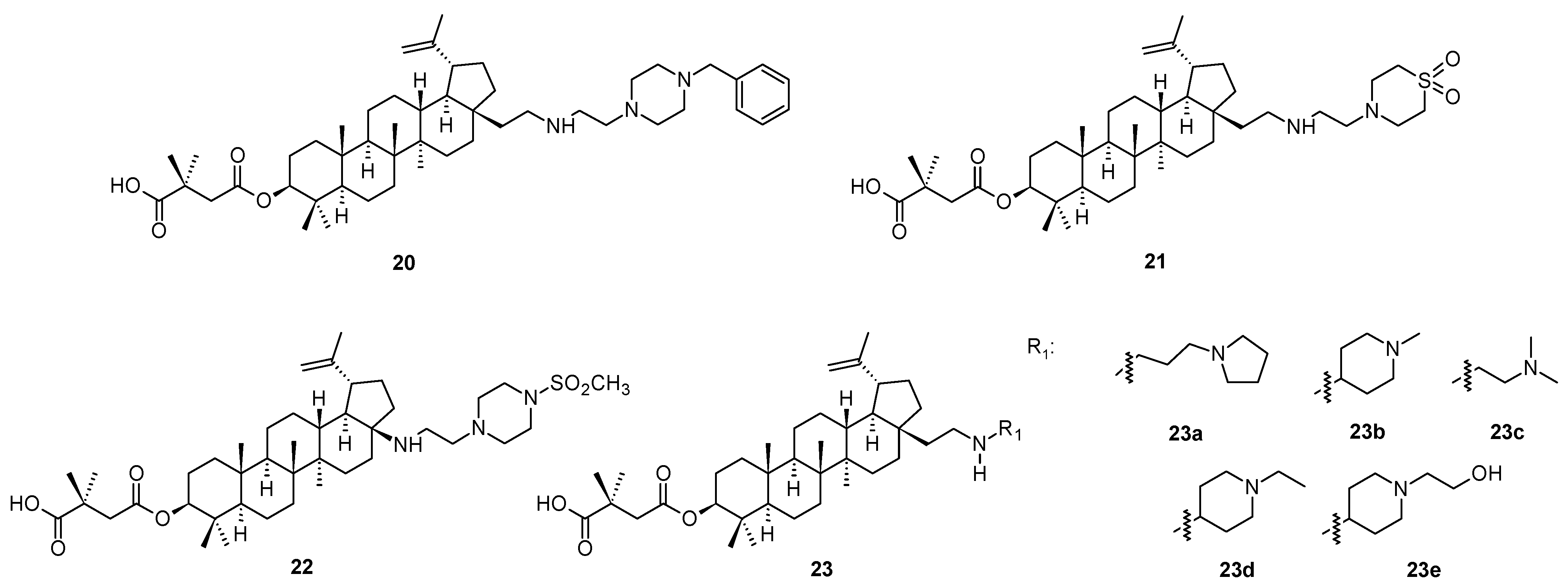
Structurally similar compounds to those mentioned above [
46,
47,
48] were reported recently as second-generation maturation inhibitors (compounds
20–
22 and
23a–
23e;
Figure 4), displaying effect higher than bevirimat (
3) against HIV-1 subtype C [
49]. In silico studies on the interaction of bevirimat and their analogs have been limited to HIV-1 subtype B (5I4T) due to the lack of an available 3D structure for HIV-1 subtype C virus. The authors [
49] have developed a 3D model of the HIV-1C Gag CA-SP1 region using protein homology modeling with HIV-1 subtype B (514T) as a template. The generated HIV-1 C homology model was extensively validated using several tools and served as a template to perform molecular docking studies with eight well-characterized maturation inhibitors. The docked complex of HIV-1C and the studied maturation inhibitors were subjected to molecular dynamics simulation for 100 ns. Based on the obtained data, it was revealed that the investigation was probably a pioneering report on the construction and validation of a 3D model for the HIV-1C Gag CA-SP1, which could serve as a crucial tool in the structure-aided design of novel and well-acting maturation inhibitors [
49]. The docking studies confirmed that modifications at the C-28 carbon centers in bevirimat analogs resulted in increased interactions with HIV-1C Gag CA-SP1 and higher binding energy as compared to the parent bevirimat (
3), which may have conferred antiviral activity to these analogs [
49]. The authors [
49] presented no antiviral activity data, however, the in silico investigation brought a novel motivation in designing more effective antiviral agents for the next generations. However, antiviral activity data of several compounds of the investigated series can be found in [
46].
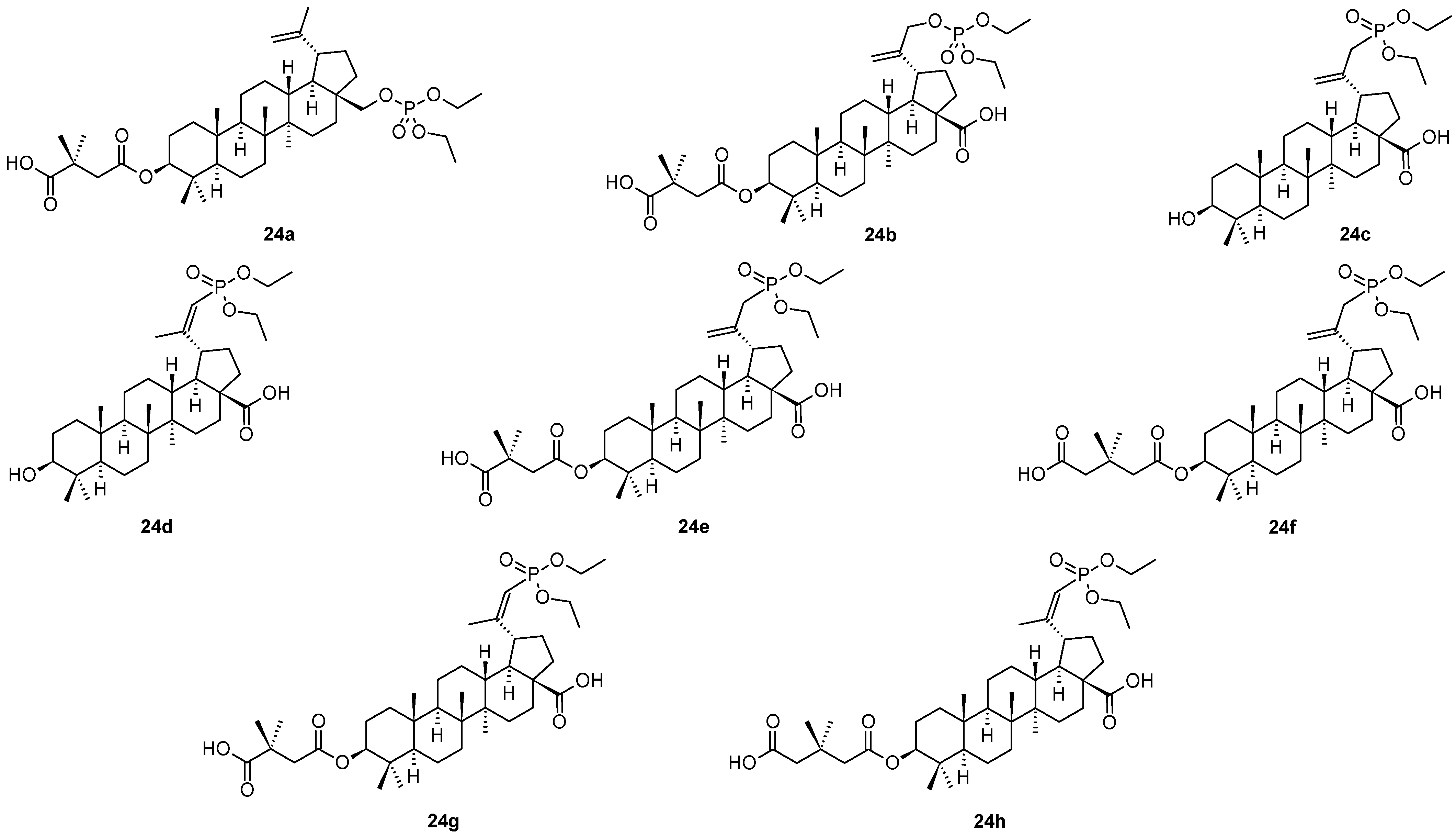
Phosphate and phosphonate derivatives of betulin (
1), betulinic acid (
2) and bevirimat (
3) represent other types of antiviral compounds displaying better pharmacological characteristics than the parent compounds [
1,
50]. Thus, several compounds of that series (
24a–
24h) are shown in
Figure 5, and their antiviral activity values are summarized in
Table 6. The inhibitory effect of
24a (
Figure 5,
Table 6) was of high value, as well as showed high therapeutic index (IC
50 = 0.02 µM, TI = 1250) on viral replication, and it displayed high selectivity [
1]. The capsid protein (CA) CTD-SP1 might be the target of
24a against HIV. Among additional phosphate and phosphonate derivatives of bevirimat (
24b–
24h), compound
24e showed antiviral activity comparable with that of
24a, however, with a slightly worse therapeutic profile than displayed by
24a.
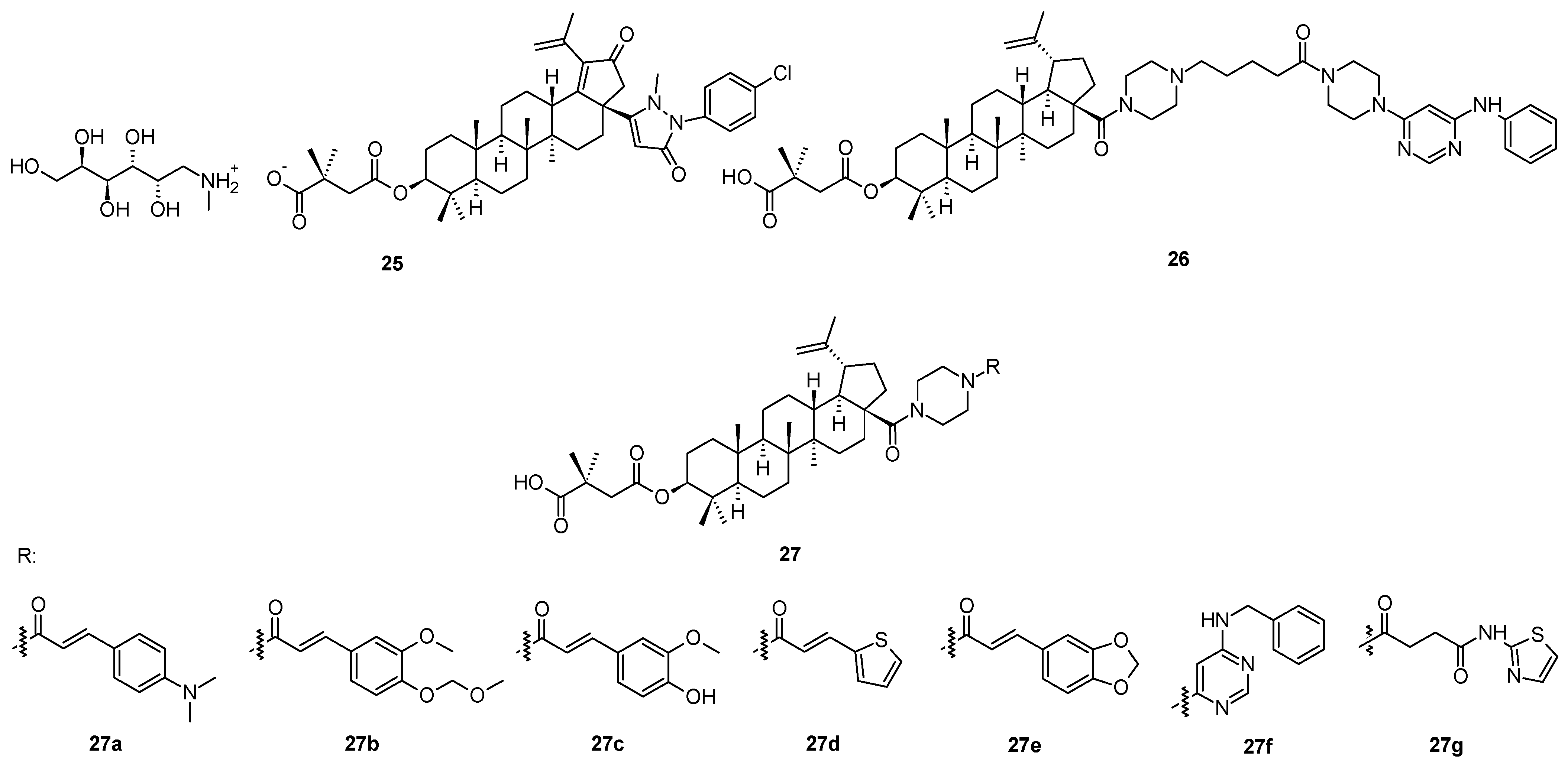
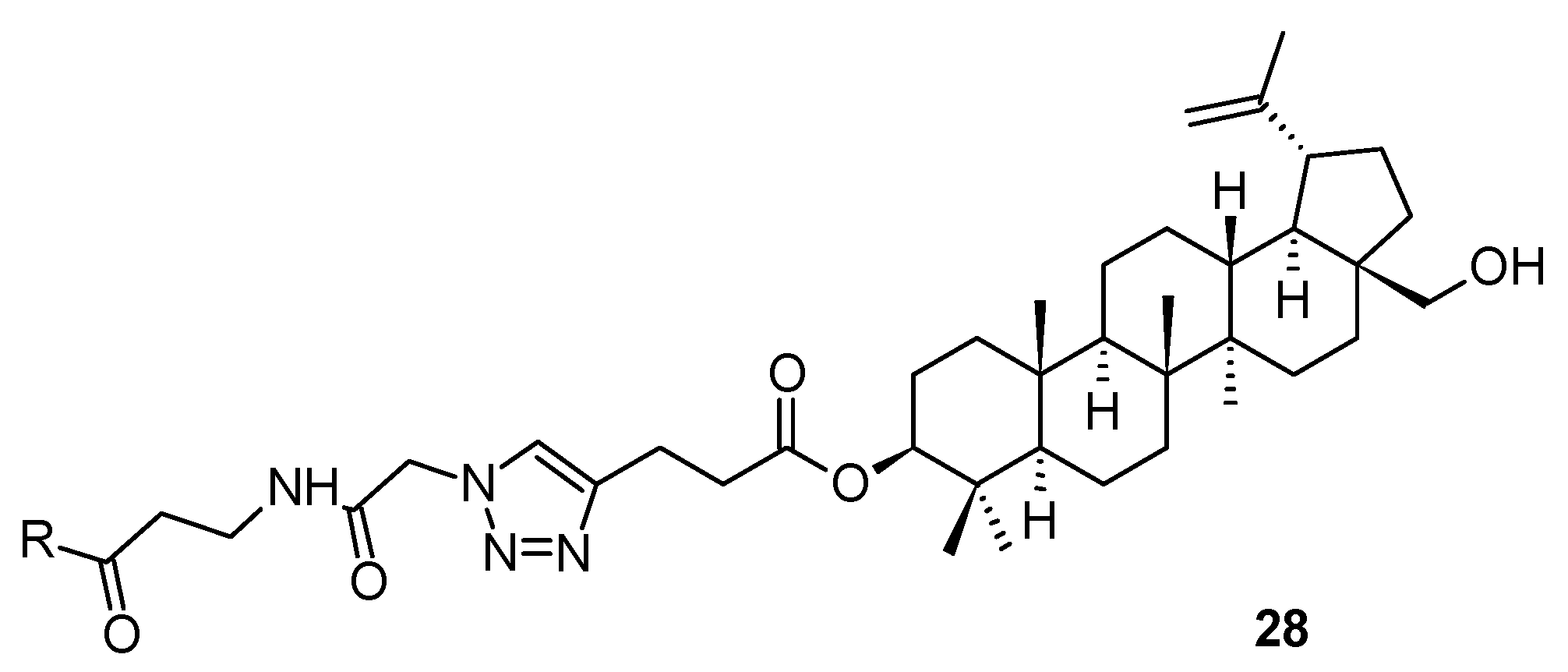
A novel compound
25 (
Figure 6), in principle, also derived from bevirimat (
3), bearing a pyrazolone system in the molecule, has been considered to represent a HIV-1 maturation inhibitor of the third generation [
51]. It displayed a maturation inhibition effect in HIV-1 with the EC
50 = 20.36 ± 2.85 nM [
51]. The mechanism of action of
25 is identical to that of the first-generation antiviral maturation inhibitor bevirimat (
3). However, the investigation showed that
25 displayed better antiviral potential than bevirimat (
3) among the virus strains tested, regardless of the presence or absence of human serum [
51]. Further designing and developing of suitable molecules resulted in a synthesis of a compound bearing selected, often called “privileged”, structural motifs (
Figure 6) [
52]. So far the most successful structure
26 (
Figure 6) showed high antiviral activity in HIV-1 NL4-3 (EC
50 = 0.012 µM), which is higher than the antiviral activity of
25. Within that series of novel piperazine-based compounds (
27a–
27g;
Figure 6), none of them showed a better antiviral effect and therapeutic profile than
26 (
Table 7).
2.3. Miscellaneous Plant Triterpenoids
Investigation of other plant triterpenoids than those of the lupane family can be found in the literature, nevertheless, it is less frequent than could be expected, even if different plant triterpenoids have been reported to display antiviral effects [
33].
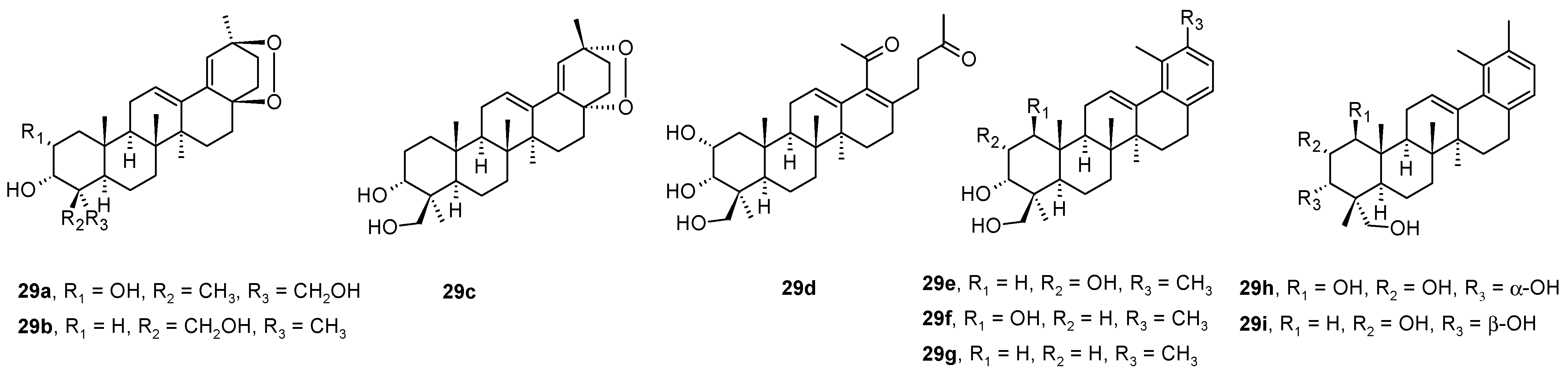
Ursane-type triterpenoids and 28-nortriterpenoids
29a−
29i (
Figure 8) were isolated from
Rhododendron latoucheae [
54]. A hyphenated NMR technique (analytical HPLC (high performance liquid chromatography) with a DAD (diode array detector) connected to MS (mass spectrometer), SPE (solid phase extraction), and NMR) has proven effective for the full structural analysis and identification of isolated natural products in complex mixtures [
54]. Compounds
29a and
29i inhibited HSV-1 in Vero cells with IC
50 values of 6.4 μM and 0.4 μM, respectively, while the compounds
29b–
29h were less effective (
Table 10) [
54].
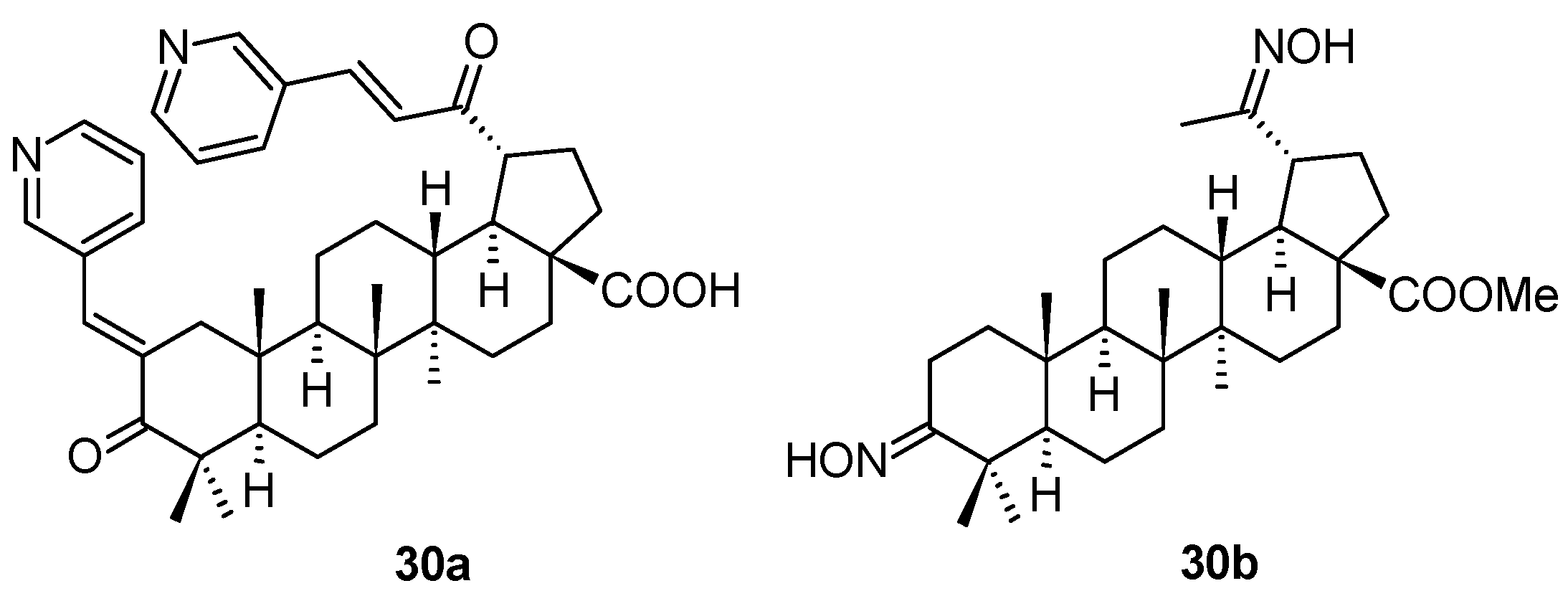
The chalcone derivatives of 20-oxo-lupanes have been synthesized and screened for several types of biological activity by Russian authors [
55]. Investigating the antiviral activity of the prepared series of compounds, two of them,
30a and
30b (
Figure 9), were evaluated as compounds displaying anti-HSV-1 activity (
Table 11). The antiviral activity of
30a and
30b was tested against HCMV, and that of
30b also against HSV-1 and HPV.
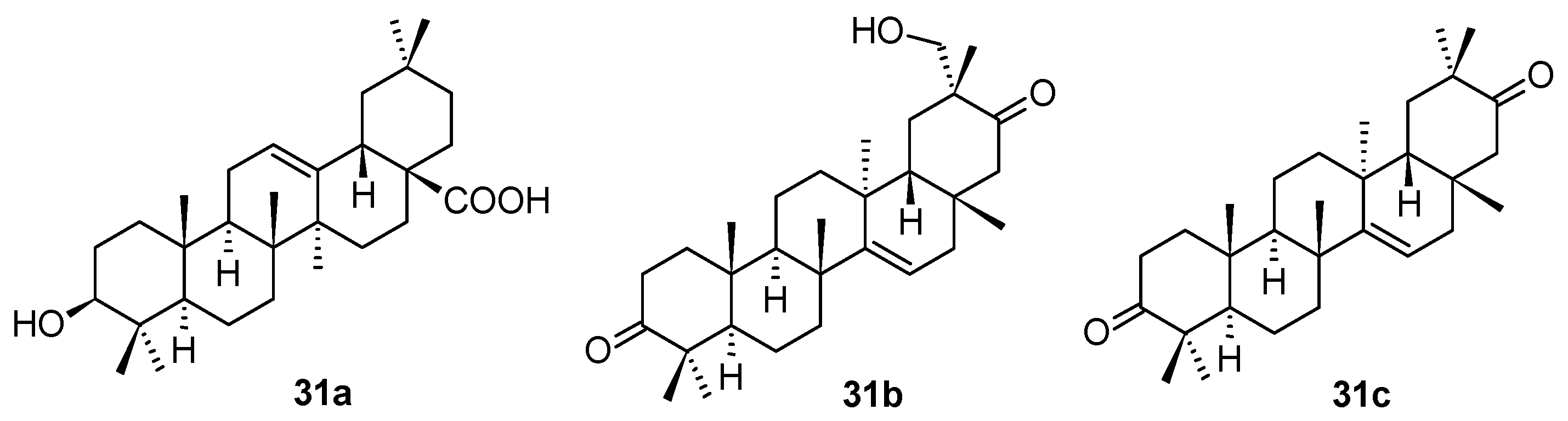
A large series of new pentacyclic triterpenoids, including oleanane-type, ursane-type, and taraxerane-type, were isolated from the stems and branches of
Enkianthus chinensis [
56]. Their structures were elucidated by extensive spectroscopic analyses, X-ray crystallographic data, and electronic circular dichroism (ECD) techniques. The in vitro biological activity evaluation resulted in a finding that the three compounds (
31a–
31c;
Figure 10) showed antiviral activity. The most active compound of this small series of natural triterpenoids was
31c showing latent activity against HSV-1 with an IC
50 value of 6.4 μM. Their structures and antiviral activity values (in comparison with those of acyclovir used as the positive reference agent) are summarized in
Table 12.
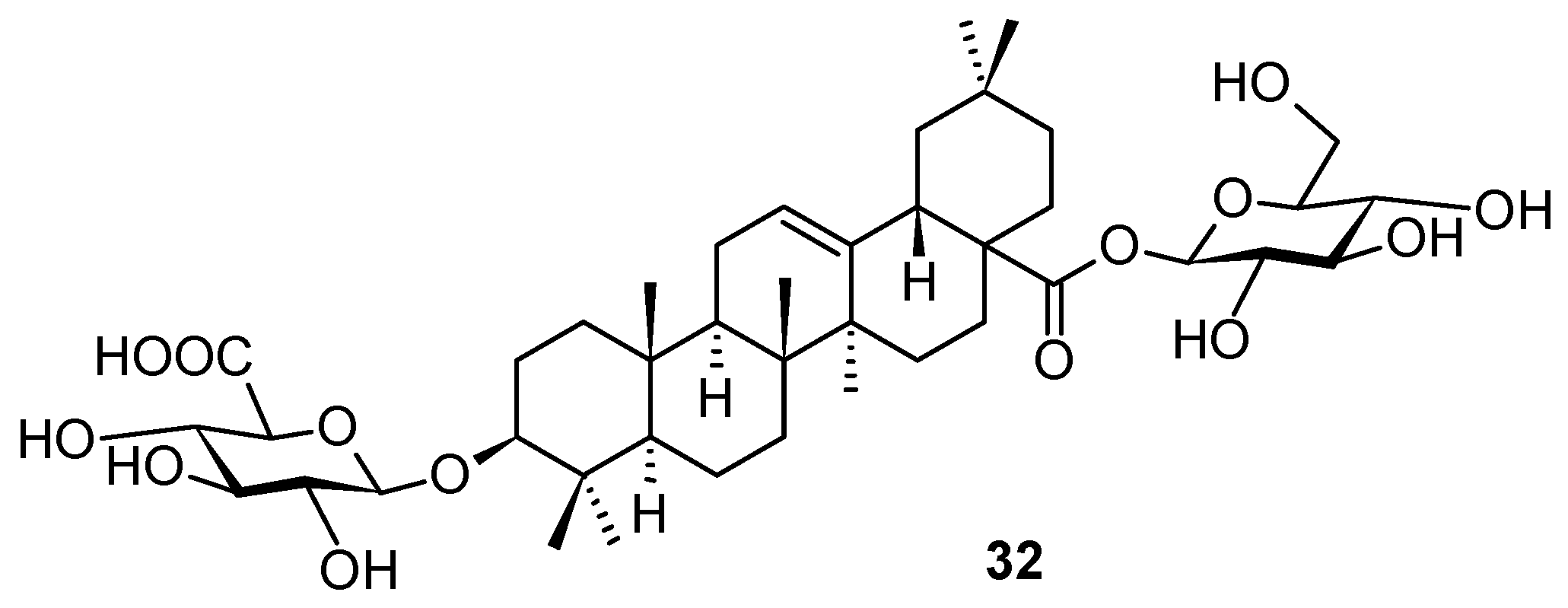
Even if triterpenoids and their natural saponin derivatives exhibit anti-HSV-1 activity, there has still been a lack of comprehensive information on the anti-HSV-1 activity of triterpenoids. Therefore, expanding information on the anti-HSV-1 activity of triterpenes and improving the efficiency of their exploration are urgently required. To improve the efficiency of the development of anti-HSV-1 active compounds, recently, Japanese authors constructed a predictive model for the anti-HSV-1 activity of triterpenes by using the information obtained from previous studies [
57]. They constructed a binary classification model (i.e., active or inactive) using a logistic regression algorithm. As a result, the assay was performed on 20 triterpenes and triterpenoids, finally identifying the structure
32 (
Figure 11) as a potent anti-HSV-1 compound, displaying IC
50 = 13.06 μM.
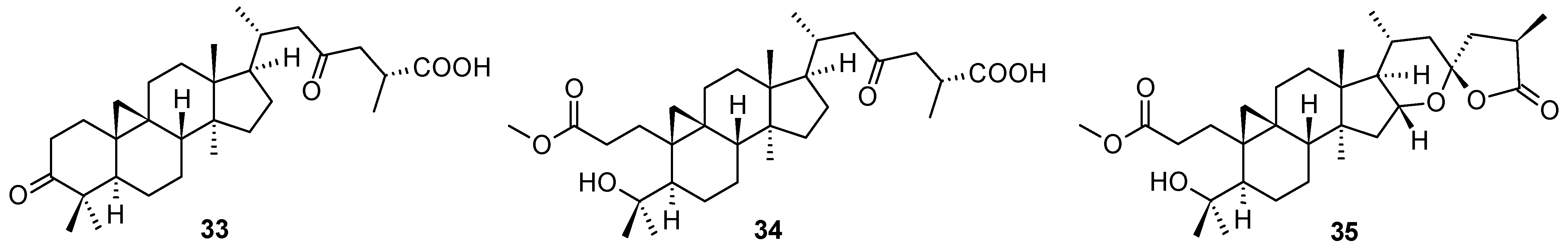
Several so far undescribed cycloartane triterpenoids, pseudolarnoids A−G, together with other known triterpenoids, were isolated from the seeds of
Pseudolarix amabilis (J. Nelson) Rehder [
58]. Their structures were elucidated on the basis of spectroscopic analysis, X-ray crystallography, and ECD data. Three of these natural products (
33–
35;
Figure 12) proved their ability to display potent antiviral effects on HSV-1 in vitro (
Table 13). Based on the therapeutic index values, structures
34 and
35 showed a better therapeutic profile than
33 (
Table 13). The structures of other less active or inactive cycloartane triterpenoids are not shown, they can be found in the original paper [
58].
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}