
Stereoselective Synthesis and Antiproliferative Activity of Steviol-Based Diterpene 1,3-Aminoalcohol Regioisomers
 , , , and
, , , and 
Abstract
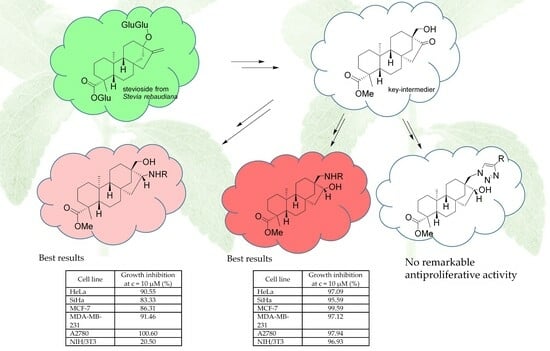
:
1. Introduction
2. Results and Discussion
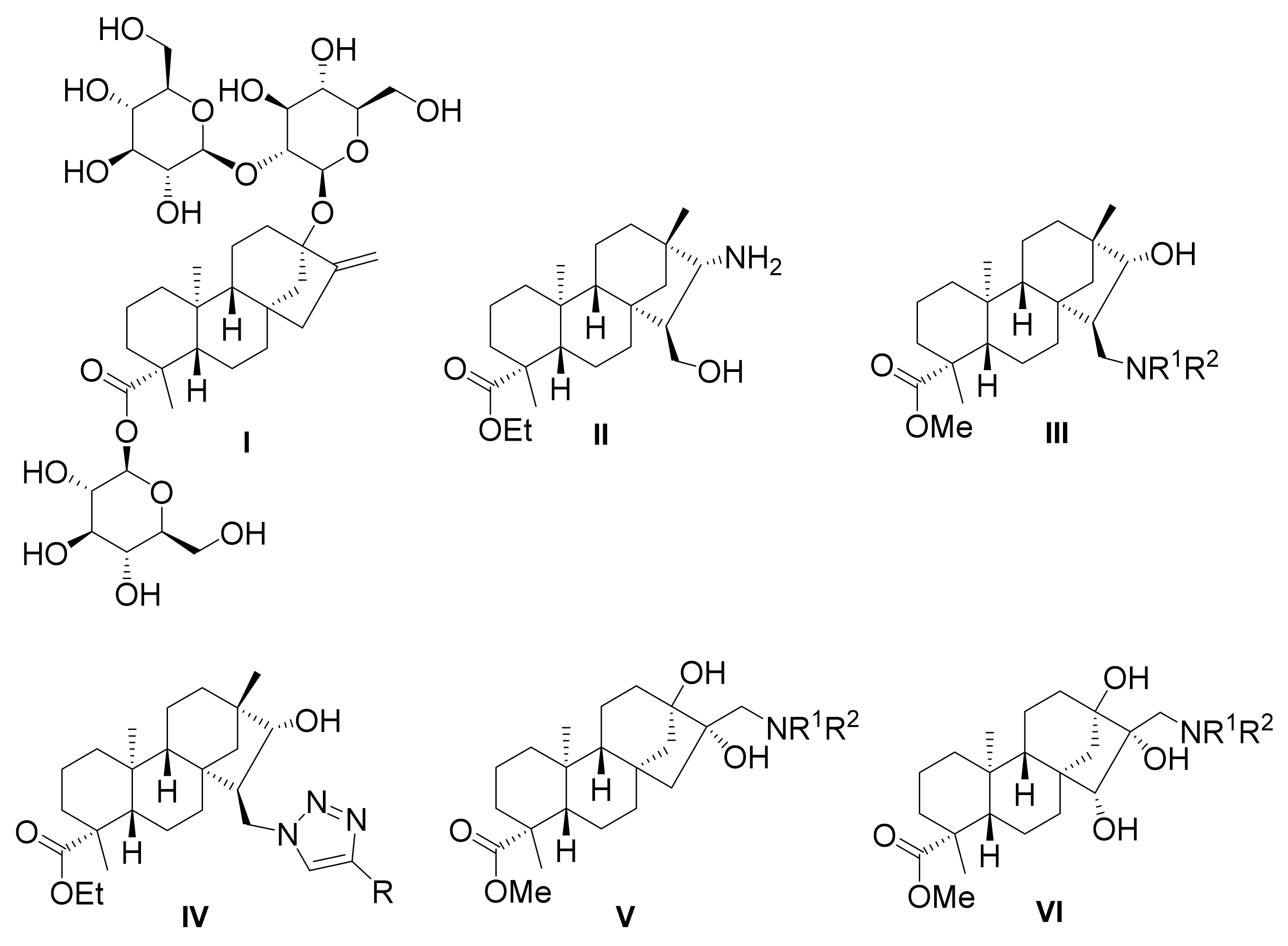
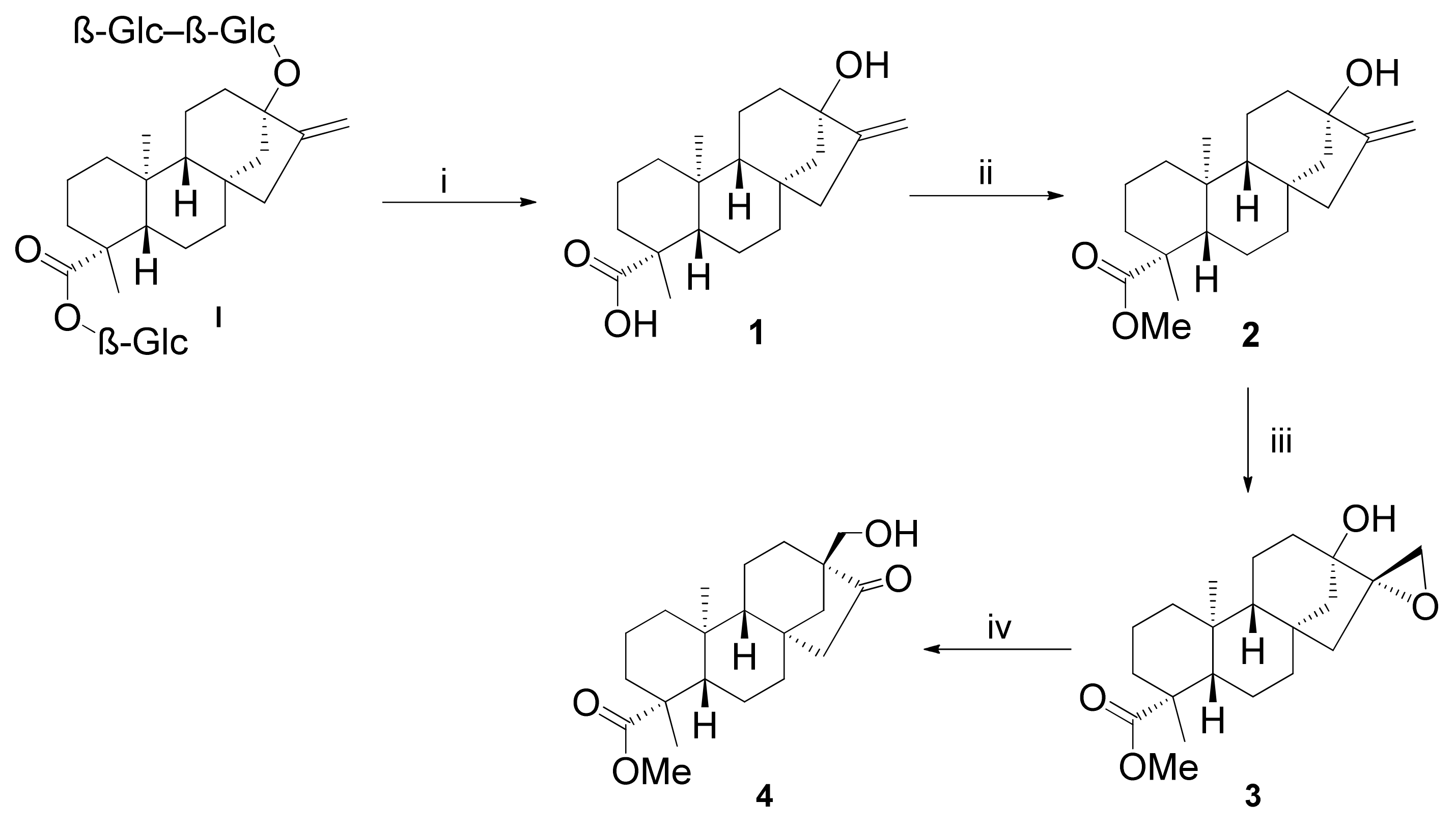
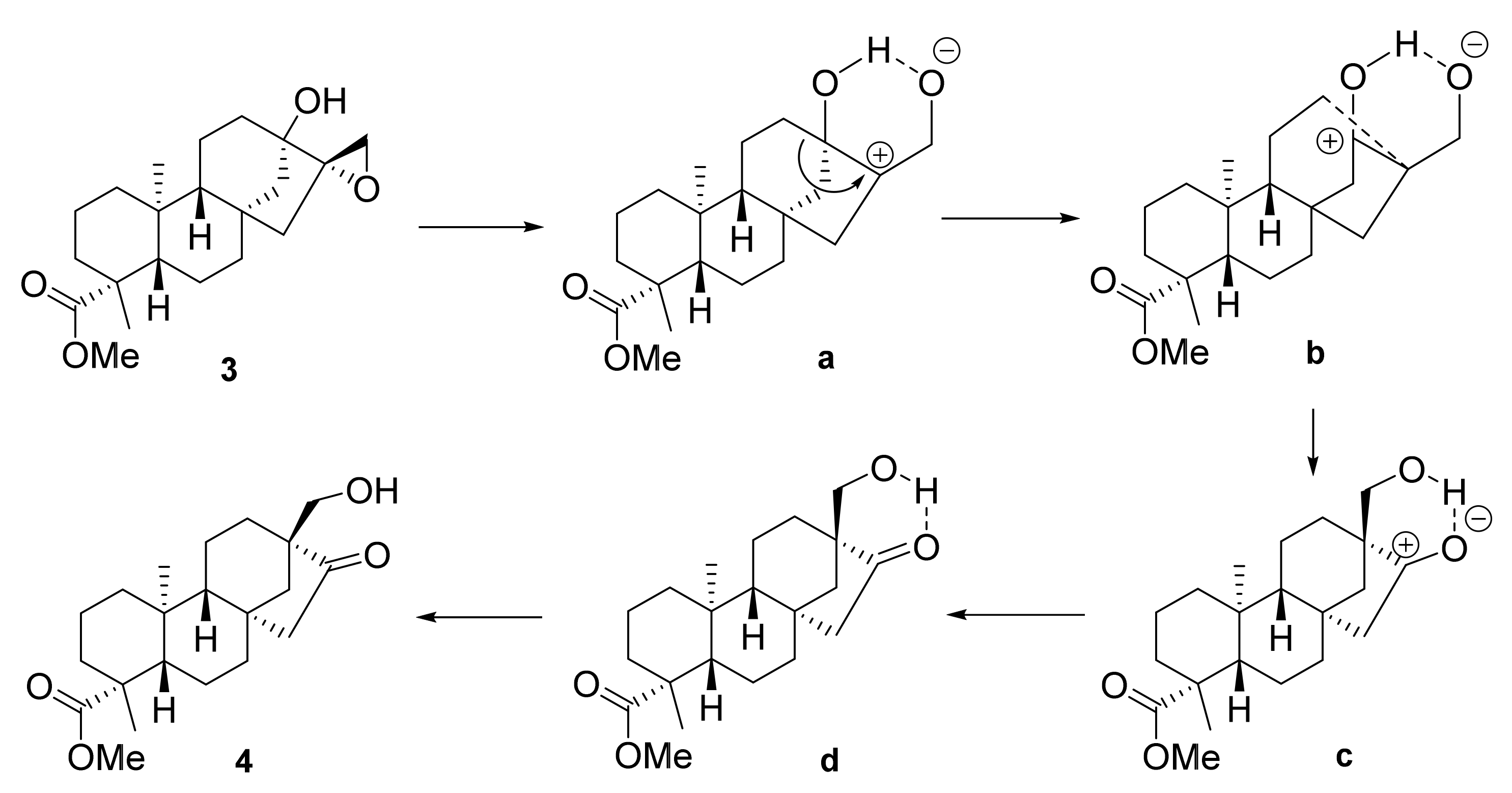
2.1. Synthesis of Key Intermediate β-Keto Alcohol 4
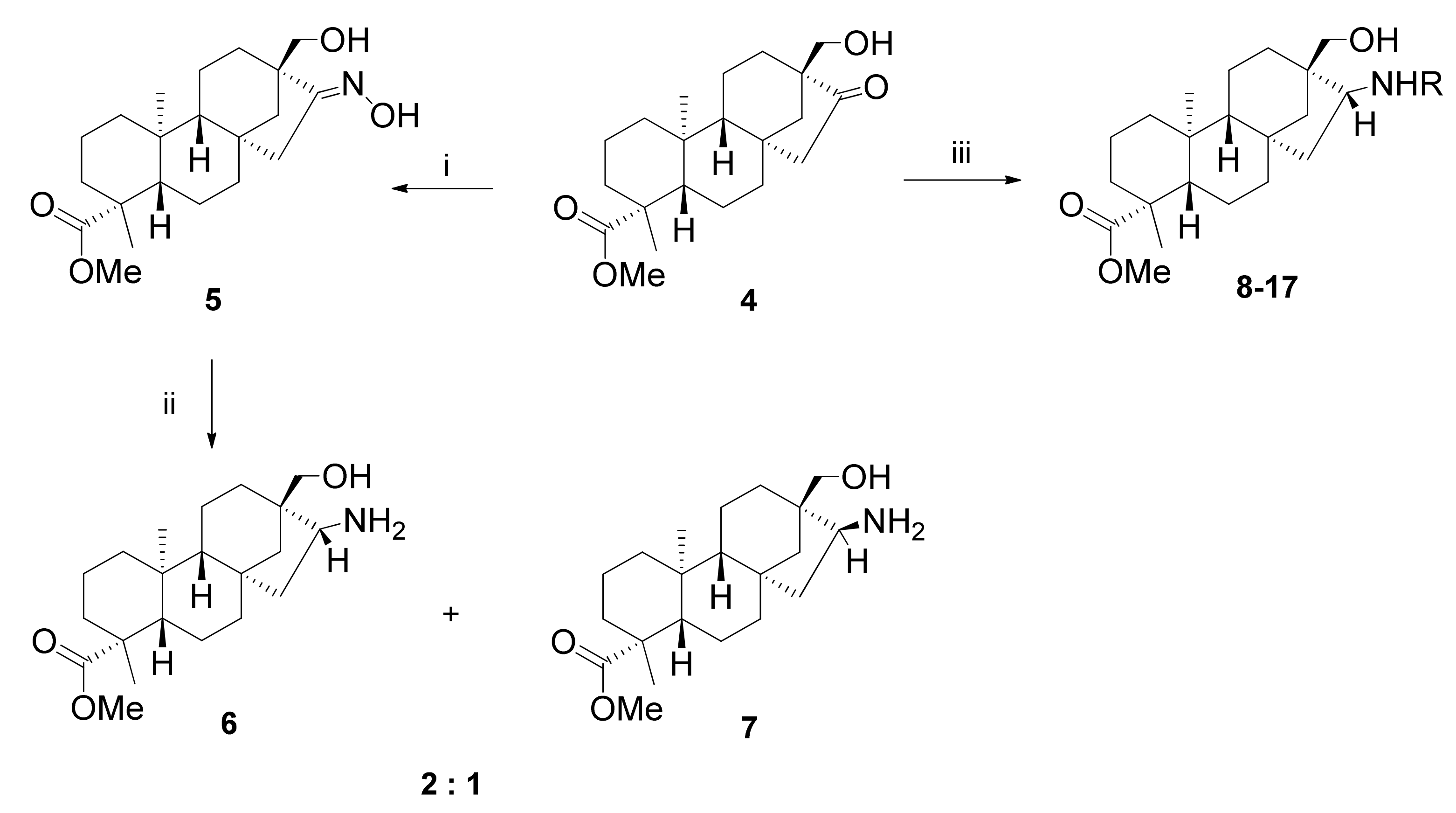
2.2. Synthesis of 1,3-Aminoalcohol Derivatives 6–17
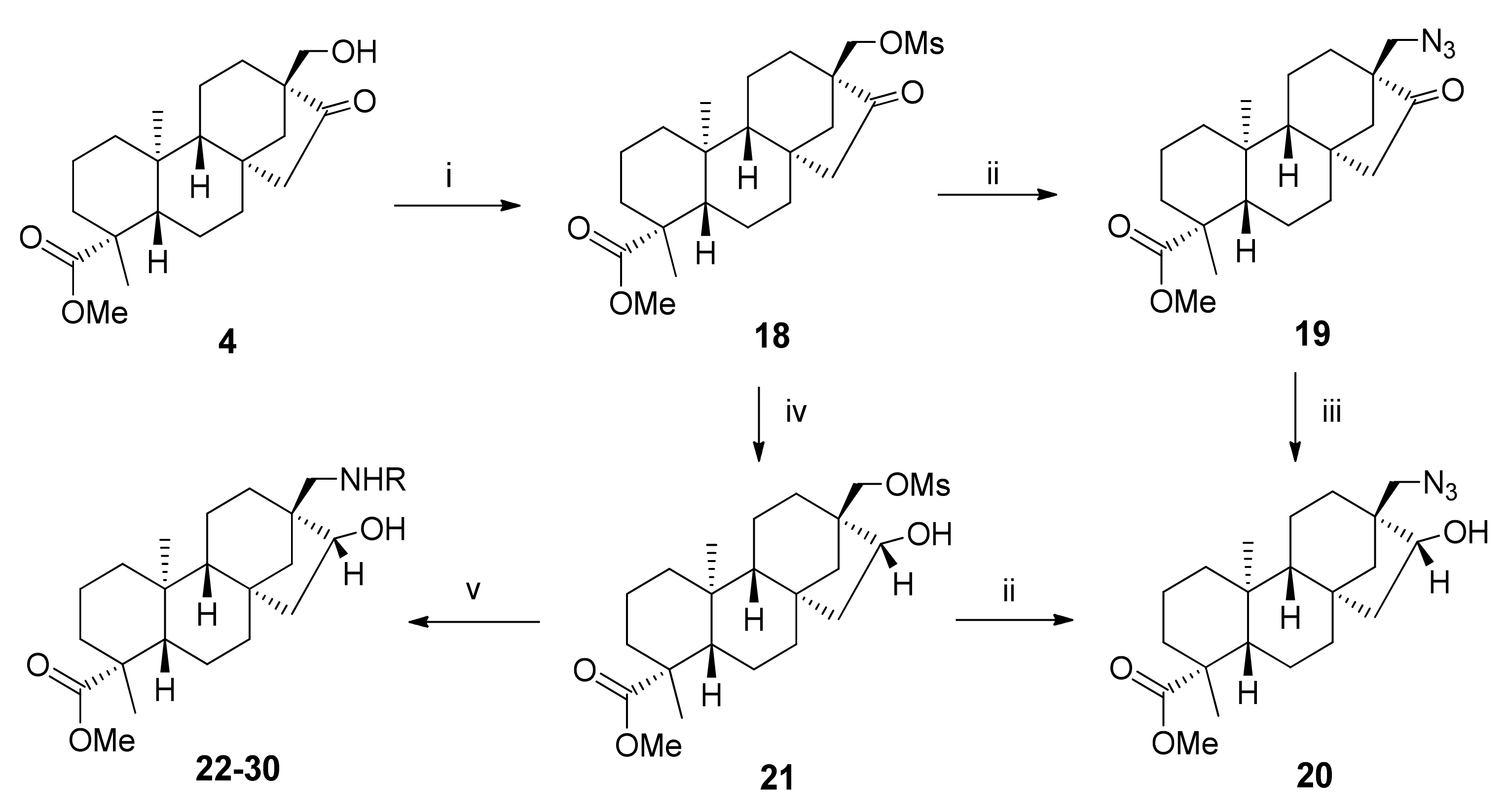
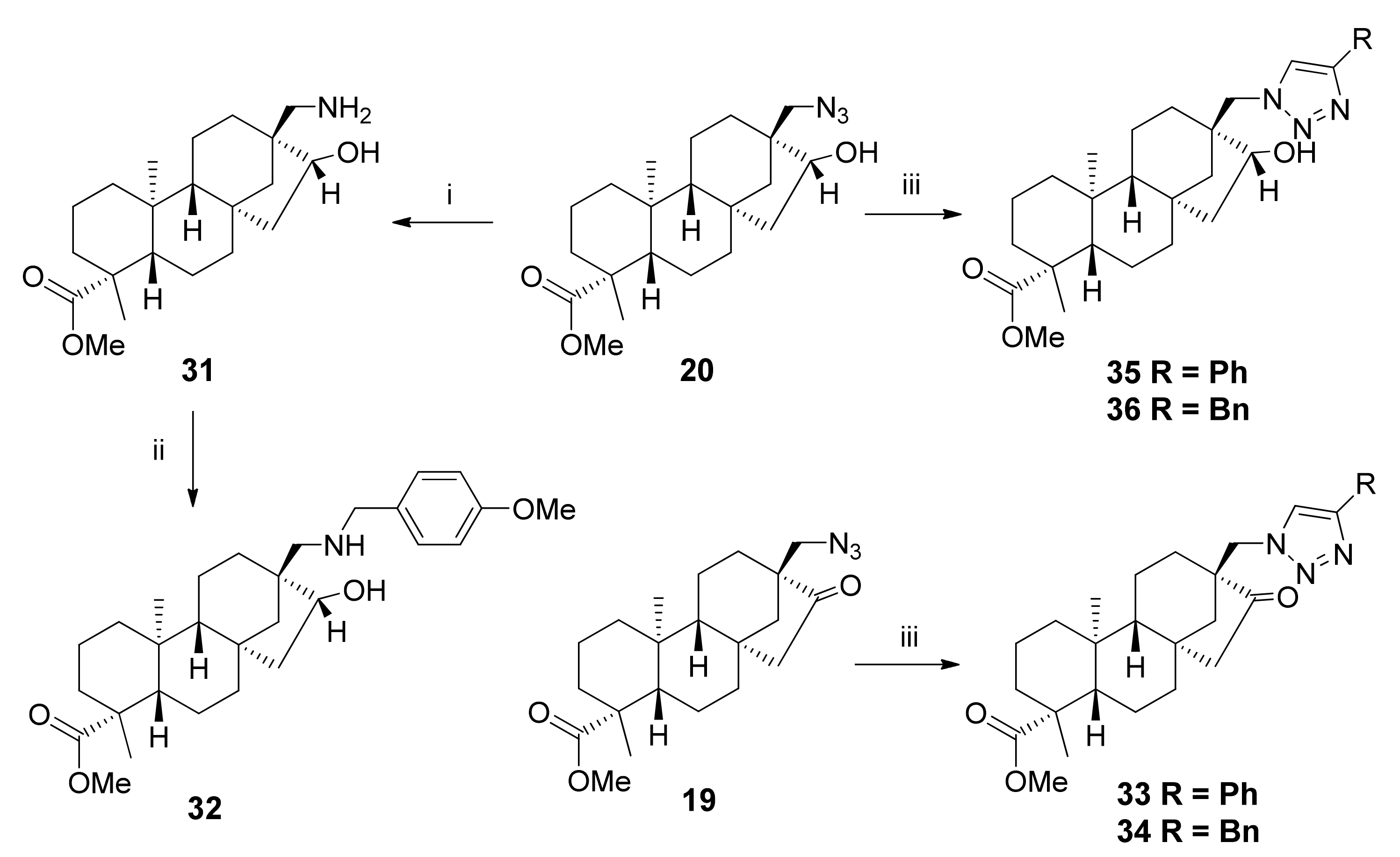
2.3. Synthesis of 1,3-Aminoalcohol Regioisomers 22–30 and Preparation of 1,2,3-Triazole Derivatives via a Click Reaction
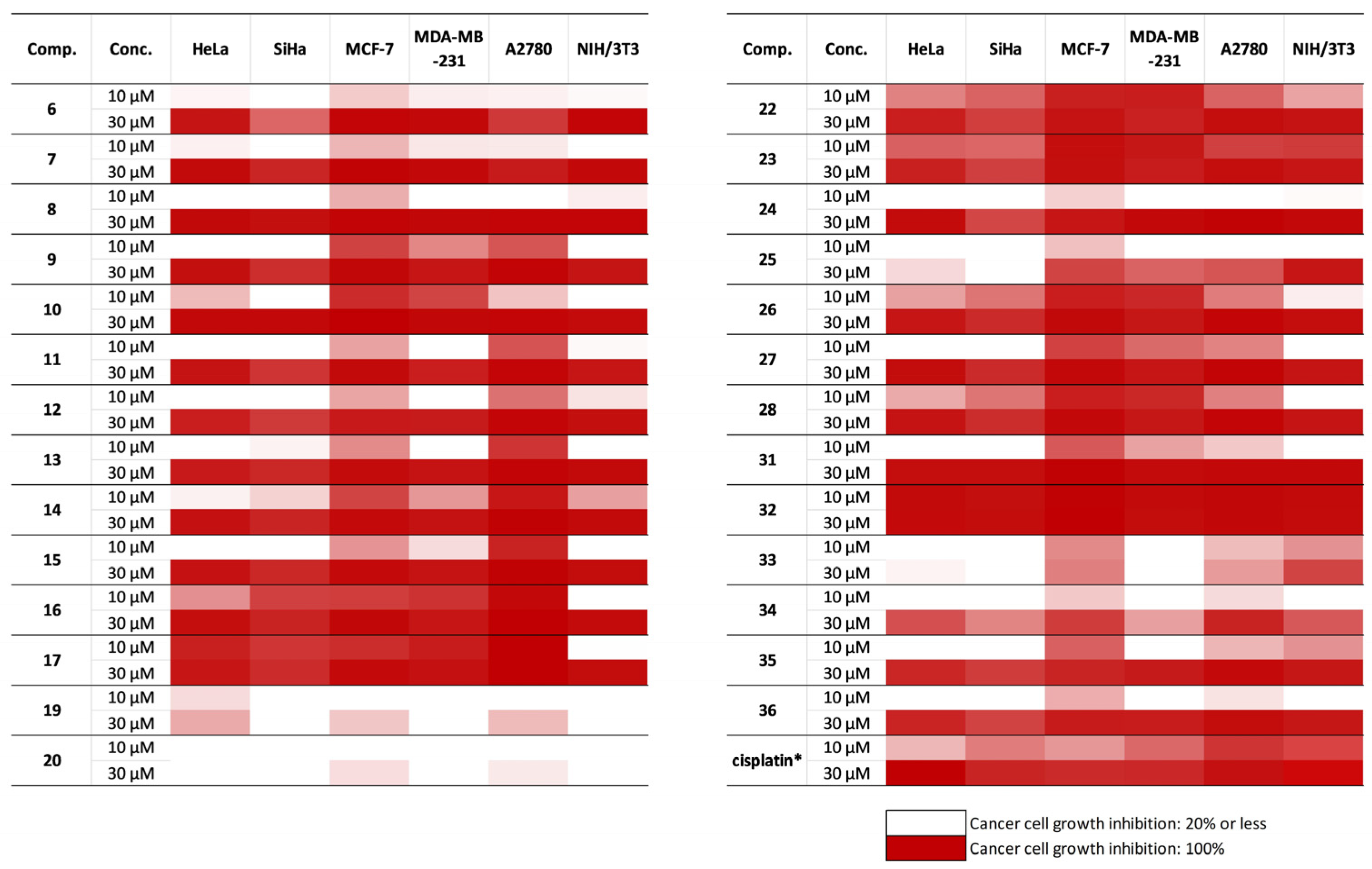
2.4. In Vitro Antiproliferative Studies of Steviol-Based Aminoalcohols and Structure–Activity Relationship
3. Materials and Methods
3.1. General Methods
3.2. Starting Materials
3.2.1. (2’S,4R,4aS,6aS,11aR,11bS)-Methyl 9-hydroxy-4,11b-dimethyldodecahydro-1H-spiro [6a,9-methanocyclohepta[a]naphthalene-8,2’-oxirane]-4-carboxylate (3)
3.2.2. (4R,4aS,6aR,9S,11aR,11bS)-Methyl 9-(hydroxymethyl)-4,11b-dimethyl-8-oxotetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (4)
3.2.3. (4R,4aS,6aR,9R,11aR,11bS,E)-Methyl 8-(hydroxyimino)-9-(hydroxymethyl)-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (5)
3.3. Hydrogenation of Oxime 5
3.3.1. (4R,4aS,6aR,8R,9R,11aR,11bS)-Methyl 8-amino-9-(hydroxymethyl)-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (6)
3.3.2. (4R,4aS,6aR,8S,9R,11aR,11bS)-Methyl 8-amino-9-(hydroxymethyl)-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (7)
3.4. General Procedure for the Preparation of Aminoalcohols Using Primary Amines from β-keto Alcohol 4
3.4.1. (4R,4aS,6aR,8R,9R,11aR,11bS)-Methyl 8-(benzylamino)-9-(hydroxymethyl)-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (8)
3.4.2. (4R,4aS,6aR,8R,9R,11aR,11bS)-Methyl 8-((4-fluorobenzyl)amino)-9-(hydroxymethyl)-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (9)
3.4.3. (4R,4aS,6aR,8R,9R,11aR,11bS)-Methyl 9-(hydroxymethyl)-8-((4-methoxybenzyl)amino)-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (10)
3.4.4. (4R,4aS,6aR,8R,9R,11aR,11bS)-Methyl 9-(hydroxymethyl)-4,11b-dimethyl-8-(((S)-1-phenylpropyl)amino)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (11)
3.4.5. (4R,4aS,6aR,8R,9R,11aR,11bS)-Methyl 9-(hydroxymethyl)-4,11b-dimethyl-8-(((R)-1-phenylpropyl)amino)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (12)
3.4.6. (4R,4aS,6aR,8R,9R,11aR,11bS)-Methyl 9-(hydroxymethyl)-4,11b-dimethyl-8-(((R)-1-(naphthalen-1-yl)ethyl)amino)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (13)
3.4.7. (4R,4aS,6aR,8R,9R,11aR,11bS)-Methyl 9-(hydroxymethyl)-4,11b-dimethyl-8-(((S)-1-(naphthalen-1-yl)ethyl)amino)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (14)
3.4.8. (4R,4aS,6aR,8R,9R,11aR,11bS)-Methyl 9-(hydroxymethyl)-4,11b-dimethyl-8-((naphthalen-1-ylmethyl)amino)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (15)
3.4.9. (4R,4aS,6aR,8R,9R,11aR,11bS)-Methyl 9-(hydroxymethyl)-4,11b-dimethyl-8-(((S)-1-(naphthalen-2-yl)ethyl)amino)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (16)
3.4.10. (4R,4aS,6aR,8R,9R,11aR,11bS)-Methyl 9-(hydroxymethyl)-4,11b-dimethyl-8-(((R)-1-(naphthalen-2-yl)ethyl)amino)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (17)
3.5. (4R,4aS,6aR,9S,11aR,11bS)-Methyl 4,11b-dimethyl-9-(((methylsulfonyl)oxy)methyl)-8-oxotetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (18)
3.6. (4R,4aS,6aR,8R,9S,11aR,11bS)-Methyl 8-hydroxy-4,11b-dimethyl-9-(((methylsulfonyl)oxy)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (21)
3.7. General Procedure for Azide Synthesis of 18 and 21 for Preparation of 19 and 20
3.7.1. (4R,4aS,6aR,9S,11aR,11bS)-Methyl 9-(azidomethyl)-4,11b-dimethyl-8-oxotetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (19)
3.7.2. (4R,4aS,6aR,8R,9S,11aR,11bS)-Methyl 9-(azidomethyl)-8-hydroxy-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (20)
3.8. General Procedure for Preparation of Aminoalcohol Regioisomers with Primary Amines from Hydroxy-Mesylate 21
3.8.1. (4R,4aS,6aR,8R,9S,11aR,11bS)-Methyl 9-((benzylamino)methyl)-8-hydroxy-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (22)
3.8.2. (4R,4aS,6aR,8R,9S,11aR,11bS)-Methyl 9-(((4-fluorobenzyl)amino)methyl)-8-hydroxy-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (23)
3.8.3. (4R,4aS,6aR,8R,9S,11aR,11bS)-Methyl 8-hydroxy-4,11b-dimethyl-9-((((R)-1-phenylpropyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (24)
3.8.4. (4R,4aS,6aR,8R,9S,11aR,11bS)-Methyl 8-hydroxy-4,11b-dimethyl-9-((((S)-1-phenylpropyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (25)
3.8.5. (4R,4aS,6aR,8R,9S,11aR,11bS)-Methyl 8-hydroxy-4,11b-dimethyl-9-(((naphthalen-1-ylmethyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (26)
3.8.6. (4R,4aS,6aR,8R,9S,11aR,11bS)-Methyl 8-hydroxy-4,11b-dimethyl-9-((((S)-1-(naphthalen-1-yl)ethyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (27)
3.8.7. (4R,4aS,6aR,8R,9S,11aR,11bS)-Methyl 8-hydroxy-4,11b-dimethyl-9-((((R)-1-(naphthalen-1-yl)ethyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (28)
3.8.8. (4R,4aS,6aR,8R,9S,11aR,11bS)-Methyl 8-hydroxy-4,11b-dimethyl-9-((((R)-1-(naphthalen-2-yl)ethyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (29)
3.8.9. (4R,4aS,6aR,8R,9S,11aR,11bS)-Methyl 8-hydroxy-4,11b-dimethyl-9-((((S)-1-(naphthalen-2-yl)ethyl)amino)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (30)
3.9. (4. R,4aS,6aR,8R,9S,11aR,11bS)-Methyl 9-(aminomethyl)-8-hydroxy-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (31)
3.10. (4R,4aS,6aR,8R,9S,11aR,11bS)-Methyl 8-hydroxy-9-(((4-methoxybenzyl)amino) methyl)-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (32)
3.11. General Procedure for Click Reaction of 19 and 20 for the Preparation of 33–36
3.11.1. (4R,4aS,6aR,9S,11aR,11bS)-Methyl 4,11b-dimethyl-8-oxo-9-((4-phenyl-1H-1,2,3-triazol-1-yl)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (33)
3.11.2. (4R,4aS,6aR,9S,11aR,11bS)-Methyl 9-((4-benzyl-1H-1,2,3-triazol-1-yl)methyl)-4,11b-dimethyl-8-oxotetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (34)
3.11.3. (4R,4aS,6aR,8R,9S,11aR,11bS)-Methyl 8-hydroxy-4,11b-dimethyl-9-((4-phenyl-1H-1,2,3-triazol-1-yl)methyl)tetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (35)
3.11.4. (4R,4aS,6aR,8R,9S,11aR,11bS)-Methyl 9-((4-benzyl-1H-1,2,3-triazol-1-yl)methyl)-8-hydroxy-4,11b-dimethyltetradecahydro-6a,9-methanocyclohepta[a]naphthalene-4-carboxylate (36)
3.12. Determination of the Antiproliferative Activities
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tetali, S.D. Terpenes and Isoprenoids: A Wealth of Compounds for Global Use. Planta 2019, 249, 1–8. [Google Scholar] [CrossRef] [PubMed]
- El Alami, M.S.I.; El Amrani, M.A.; Agbossou-Niedercorn, F.; Suisse, I.; Mortreux, A. Chiral Ligands Derived from Monoterpenes: Application in the Synthesis of Optically Pure Secondary Alcohols via Asymmetric Catalysis. Chem. Eur. J. 2015, 21, 1398–1413. [Google Scholar] [CrossRef] [PubMed]
- De las Casas Engel, T.; Maroto, B.L.; Martínez, A.G.; de la Moya Cerero, S. N/N/O versus N/O/O and N/O Amino Isoborneols in the Enantioselective Ethylation of Benzaldehyde. Tetrahedron Asymmetry 2008, 19, 269–272. [Google Scholar] [CrossRef]
- Sánchez-Carnerero, E.M.; de las Casas Engel, T.; Maroto, B.L.; de la Moya Cerero, S. Polyoxygenated Ketopinic-Acid-Derived γ-Amino Alcohols in the Enantioselective Diethylzinc Addition to Benzaldehyde. Tetrahedron Asymmetry 2009, 20, 2655–2657. [Google Scholar] [CrossRef]
- Jaworska, M.; Błocka, E.; Kozakiewicz, A.; Wełniak, M. α-Pinene-Type Chiral Schiff Bases as Tridentate Ligands in Asymmetric Addition Reactions. Tetrahedron Asymmetry 2011, 22, 648–657. [Google Scholar] [CrossRef]
- Evans, P.A.; Brandt, T.A. Enantioselective Allylic Substitution Using a Novel (Phosphino-1,3-Oxazine)Palladium Catalyst. Tetrahedron Lett. 1996, 37, 9143–9146. [Google Scholar] [CrossRef]
- Szakonyi, Z.; Balázs, Á.; Martinek, T.A.; Fülöp, F. Enantioselective Addition of Diethylzinc to Aldehydes Catalyzed by γ-Amino Alcohols Derived from (+)- and (−)-α-Pinene. Tetrahedron Asymmetry 2006, 17, 199–204. [Google Scholar] [CrossRef]
- Szakonyi, Z.; Zupkó, I.; Sillanpää, R.; Fülöp, F. Stereoselective Synthesis and Cytoselective Toxicity of Monoterpene-Fused 2-Imino-1,3-Thiazines. Molecules 2014, 19, 15918–15937. [Google Scholar] [CrossRef]
- Raji, M.; Le, T.; Huynh, T.; Szekeres, A.; Nagy, V.; Zupko, I.; Szakonyi, Z. Divergent Synthesis, Antiproliferative and Antimicrobial Studies of 1,3-Aminoalcohol and 3-Amino-1,2-Diol Based Diaminopyrimidines. Chem. Biodivers. 2022, 19, e202200077. [Google Scholar] [CrossRef]
- Koneva, E.A.; Khomenko, T.M.; Kurbakova, S.Y.; Komarova, N.I.; Korchagina, D.V.; Volcho, K.P.; Salakhutdinov, N.F.; Tolstikov, A.G.; Tolstikov, G.A. Synthesis of Optically Active Omeprazole by Catalysis with Vanadyl Complexes with Chiral Schiff Bases. Russ. Chem. Bull. 2008, 57, 1680–1685. [Google Scholar] [CrossRef]
- Koneva, E.A.; Korchagina, D.V.; Gatilov, Y.V.; Genaev, A.M.; Krysin, A.P.; Volcho, K.P.; Tolstikov, A.G.; Salakhutdinov, N.F. New Chiral Ligands Based on (+)-α-Pinene. Russ. J. Org. Chem. 2010, 46, 1109–1115. [Google Scholar] [CrossRef]
- Woltering, T.J.; Wostl, W.; Hilpert, H.; Rogers-Evans, M.; Pinard, E.; Mayweg, A.; Göbel, M.; Banner, D.W.; Benz, J.; Travagli, M.; et al. BACE1 Inhibitors: A Head Group Scan on a Series of Amides. Bioorg. Med. Chem. Lett. 2013, 23, 4239–4243. [Google Scholar] [CrossRef] [PubMed]
- Dragomanova, S.; Andonova, V.; Volcho, K.; Salakhutdinov, N.; Kalfin, R.; Tancheva, L. Therapeutic Potential of Myrtenal and Its Derivatives—A Review. Life 2023, 13, 2086. [Google Scholar] [CrossRef] [PubMed]
- Ludwiczuk, A.; Skalicka-Woźniak, K.; Georgiev, M.I. Terpenoids. In Pharmacognosy; Elsevier: Amsterdam, The Netherlands, 2017; pp. 233–266. ISBN 978-0-12-802104-0. [Google Scholar]
- Wang, S.; Rodríguez-Escrich, C.; Fan, X.; Pericàs, M.A. A Site Isolation-Enabled Organocatalytic Approach to Enantiopure γ-Amino Alcohol Drugs. Tetrahedron 2018, 74, 3943–3946. [Google Scholar] [CrossRef]
- Perrine, D.M.; Sabanayagam, N.R.; Reynolds, K.J. Synthesis of NMP, a Fluoxetine (Prozac) Precursor, in the Introductory Organic Laboratory. J. Chem. Educ. 1998, 75, 1266. [Google Scholar] [CrossRef]
- Dicpinigaitis, P.V.; Morice, A.H.; Birring, S.S.; McGarvey, L.; Smith, J.A.; Canning, B.J.; Page, C.P. Antitussive Drugs—Past, Present, and Future. Pharmacol. Rev. 2014, 66, 468–512. [Google Scholar] [CrossRef] [PubMed]
- Carocho, M.; Morales, P.; Ferreira, I.C.F.R. Sweeteners as Food Additives in the XXI Century: A Review of What Is Known, and What Is to Come. Food Chem. Toxicol. 2017, 107, 302–317. [Google Scholar] [CrossRef]
- Wang, M.; Li, H.; Xu, F.; Gao, X.; Li, J.; Xu, S.; Zhang, D.; Wu, X.; Xu, J.; Hua, H.; et al. Diterpenoid Lead Stevioside and Its Hydrolysis Products Steviol and Isosteviol: Biological Activity and Structural Modification. Eur. J. Med. Chem. 2018, 156, 885–906. [Google Scholar] [CrossRef]
- Liu, C.-J.; Yu, S.-L.; Liu, Y.-P.; Dai, X.-J.; Wu, Y.; Li, R.-J.; Tao, J.-C. Synthesis, Cytotoxic Activity Evaluation and HQSAR Study of Novel Isosteviol Derivatives as Potential Anticancer Agents. Eur. J. Med. Chem. 2016, 115, 26–40. [Google Scholar] [CrossRef]
- Ozsvár, D.; Nagy, V.; Zupkó, I.; Szakonyi, Z. Synthesis and Biological Application of Isosteviol-Based 1,3-Aminoalcohols. Int. J. Mol. Sci. 2021, 22, 11232. [Google Scholar] [CrossRef]
- Liu, C.-J.; Liu, Y.-P.; Yu, S.-L.; Dai, X.-J.; Zhang, T.; Tao, J.-C. Syntheses, Cytotoxic Activity Evaluation and HQSAR Study of 1,2,3-Triazole-Linked Isosteviol Derivatives as Potential Anticancer Agents. Bioorg. Med. Chem. Lett. 2016, 26, 5455–5461. [Google Scholar] [CrossRef] [PubMed]
- Ozsvár, D.; Nagy, V.; Zupkó, I.; Szakonyi, Z. Stereoselective Synthesis and Antiproliferative Activity of Steviol-Based Diterpen Aminodiols. Int. J. Mol. Sci. 2020, 21, 184. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Schelz, Z.; Erdős, D.; Kis, A.K.; Nagy, V.; Zupkó, I.; Balogh, G.T.; Szakonyi, Z. Stereoselective Synthesis and Antiproliferative Activities of Tetrafunctional Diterpene Steviol Derivatives. Int. J. Mol. Sci. 2023, 24, 1121. [Google Scholar] [CrossRef] [PubMed]
- Ukiya, M.; Sawada, S.; Kikuchi, T.; Kushi, Y.; Fukatsu, M.; Akihisa, T. Cytotoxic and Apoptosis-Inducing Activities of Steviol and Isosteviol Derivatives against Human Cancer Cell Lines. Chem. Biodivers. 2013, 10, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.-Y.; Wu, J.-Q.; Zhang, D.-Y.; Wu, Y.-C.; Hua, W.-Y.; Wu, X.-M. Efficient Synthesis of Novel Jolkinolides and Related Derivatives Starting from Stevioside. Synthesis 2011, 2011, 3807–3814. [Google Scholar] [CrossRef]
- Vandichel, M.; Leus, K.; Van Der Voort, P.; Waroquier, M.; Van Speybroeck, V. Mechanistic Insight into the Cyclohexene Epoxidation with VO(Acac)2 and Tert-Butyl Hydroperoxide. J. Catal. 2012, 294, 1–18. [Google Scholar] [CrossRef]
- Khaibullin, R.N.; Strobykina, I.Y.; Kataev, V.E.; Lodochnikova, O.A.; Gubaidullin, A.T.; Balandina, A.A.; Latypov, S.K. Wagner-Meerwein Rearrangement of Steviol 16α,17- and 15α,16-Epoxides. Russ. J. Org. Chem. 2010, 46, 1006–1012. [Google Scholar] [CrossRef]
- Santana, V.C.S.; Rocha, E.C.S.; Pavan, J.C.S.; Heleno, V.C.G.; De Lucca, E.C. Selective Oxidations in the Synthesis of Complex Natural Ent-Kauranes and Ent-Beyeranes. J. Org. Chem. 2022, 87, 10462–10466. [Google Scholar] [CrossRef]
- Schreiber, K.; Schneider, G.; Sembdner, G. Gibberelline—XII. Tetrahedron 1968, 24, 73–78. [Google Scholar] [CrossRef]
- De Oliveira, B.H.; Stiirmer, J.C.; De Souza Filho, J.D.; Ayub, R.A. Plant Growth Regulation Activity of Steviol and Derivatives. Phytochemistry 2008, 69, 1528–1533. [Google Scholar] [CrossRef]
- Hamada, Y.; Shioiri, T. Cycloundecanecarboxylic Acid. Org. Synth. 1984, 62, 191. [Google Scholar] [CrossRef]
- Fürst, A.; Koller, F. Über Steroide Und Sexualhormone. Ein Neuer Weg Zur Herstellung Der A-Oxyde von Cholesterin Und Trans-Dehydro-androsteron. Helv. Chim. Acta 1947, 30, 1454–1460. [Google Scholar] [CrossRef] [PubMed]
- Causero, A.; Troll, C.; Rieger, B. (+)-Limonene Functionalization: Syntheses, Optimization, and Scale-up Procedures for Sustainable Polymer Building Blocks. Ind. Eng. Chem. Res. 2020, 59, 15464–15477. [Google Scholar] [CrossRef]
- Pöhler, R.; Krahn, J.H.; van den Boom, J.; Dobrynin, G.; Kaschani, F.; Eggenweiler, H.; Zenke, F.T.; Kaiser, M.; Meyer, H. A Non-Competitive Inhibitor of VCP/P97 and VPS4 Reveals Conserved Allosteric Circuits in Type I and II AAA ATPases. Angew. Chem. Int. Ed. 2018, 57, 1576–1580. [Google Scholar] [CrossRef] [PubMed]
- Csuk, R.; Deigner, H.-P. The Potential of Click Reactions for the Synthesis of Bioactive Triterpenes. Bioorg. Med. Chem. Lett. 2019, 29, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Worrell, B.T.; Malik, J.A.; Fokin, V.V. Direct Evidence of a Dinuclear Copper Intermediate in Cu(I)-Catalyzed Azide-Alkyne Cycloadditions. Science 2013, 340, 457–460. [Google Scholar] [CrossRef] [PubMed]
- Himo, F.; Lovell, T.; Hilgraf, R.; Rostovtsev, V.V.; Noodleman, L.; Sharpless, K.B.; Fokin, V.V. Copper(I)-Catalyzed Synthesis of Azoles. DFT Study Predicts Unprecedented Reactivity and Intermediates. J. Am. Chem. Soc. 2005, 127, 210–216. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Subrahmanyam, C.; Rao, B.V.; Ward, R.S.; Hursthouse, M.B.; Hibbs, D.E. Diterpenes from the Marine Mangrove Bruguieragymnorhiza. Phytochemistry 1999, 51, 83–90. [Google Scholar] [CrossRef]
- Nanayakkara, N.P.D.; Klocke, J.A.; Compadre, C.M.; Hussain, R.A.; Pezzuto, J.M.; Kinghorn, A.D. Characterization and Feeding Deterrent Effects on the Aphid, Schizaphis Graminum, of Some Derivatives of the Sweet Compounds, Stevioside and Rebaudioside A. J. Nat. Prod. 1987, 50, 434–441. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Product | R | Yield [%] |
|---|---|---|---|
| 1 | 8 | benzyl | 43 |
| 2 | 9 | 4-fluorobenzyl | 48 |
| 3 | 10 | 4-methoxybenzyl | 70 |
| 4 | 11 | (R)-α-ethylbenzyl | 65 |
| 5 | 12 | (S)-α-ethylbenzyl | 60 |
| 6 | 13 | (R)-1-(1-naphthyl)ethyl | 41 |
| 7 | 14 | (S)-1-(1-naphthyl)ethyl | 38 |
| 8 | 15 | 1-naphthylmethyl | 78 |
| 9 | 16 | (R)-1-(2-naphthyl)ethyl | 60 |
| 10 | 17 | (S)-1-(2-naphthyl)ethyl | 56 |
| Entry | Product | R | Yield [%] |
|---|---|---|---|
| 1 | 22 | benzyl | 22 |
| 2 | 23 | 4-fluorobenzyl | 41 |
| 3 | 24 | (R)-α-ethylbenzyl | 40 |
| 4 | 25 | (S)-α-ethylbenzyl | 36 |
| 5 | 26 | 1-naphthylmethyl | 44 |
| 6 | 27 | (R)-1-(1-naphthyl)ethyl | 29 |
| 7 | 28 | (S)-1-(1-naphthyl)ethyl | 21 |
| 8 | 29 | (R)-1-(2-naphthyl)ethyl | 19 |
| 9 | 30 | (S)-1-(2-naphthyl)ethyl | 15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, D.; Schelz, Z.; Boncz, M.F.; Zupkó, I.; Szakonyi, Z. Stereoselective Synthesis and Antiproliferative Activity of Steviol-Based Diterpene 1,3-Aminoalcohol Regioisomers. Molecules 2023, 28, 7962. https://doi.org/10.3390/molecules28247962
Bai D, Schelz Z, Boncz MF, Zupkó I, Szakonyi Z. Stereoselective Synthesis and Antiproliferative Activity of Steviol-Based Diterpene 1,3-Aminoalcohol Regioisomers. Molecules. 2023; 28(24):7962. https://doi.org/10.3390/molecules28247962
Chicago/Turabian StyleBai, Dorottya, Zsuzsanna Schelz, Mária Fanni Boncz, István Zupkó, and Zsolt Szakonyi. 2023. "Stereoselective Synthesis and Antiproliferative Activity of Steviol-Based Diterpene 1,3-Aminoalcohol Regioisomers" Molecules 28, no. 24: 7962. https://doi.org/10.3390/molecules28247962
APA StyleBai, D., Schelz, Z., Boncz, M. F., Zupkó, I., & Szakonyi, Z. (2023). Stereoselective Synthesis and Antiproliferative Activity of Steviol-Based Diterpene 1,3-Aminoalcohol Regioisomers. Molecules, 28(24), 7962. https://doi.org/10.3390/molecules28247962