
Demonstration of the Formation of a Selenocysteine Selenenic Acid through Hydrolysis of a Selenocysteine Selenenyl Iodide Utilizing a Protective Molecular Cradle


Abstract
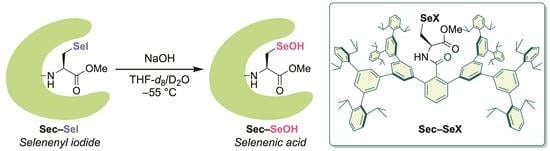
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Synthesis of Sec–SeI 4
3.3. Hydrolysis of Sec–SeI 4 and Derivatization of Resulting Sec–SeOH 2 with Dimedone (5)
3.4. X-ray Crystallographic Analysis of Sec–SeI 4
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kryukov, G.V.; Castellano, S.; Novoselov, S.V.; Lobanov, A.V.; Zehtab, O.; Guigo, R.; Gladyshev, V.N. Characterization of Mammalian Selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Rozovsky, S. Contribution of selenocysteine to the peroxidase activity of selenoprotein S. Biochemistry 2013, 52, 5514–5516. [Google Scholar] [CrossRef] [PubMed]
- Zeida, A.; Trujillo, M.; Ferrer-Sueta, G.; Denicola, A.; Estrin, D.A.; Radi, R. Catalysis of Peroxide Reduction by Fast Reacting Protein Thiols. Chem. Rev. 2019, 119, 10829–10855. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.; Lee, J.; Wu, C.; Guo, X.; Lee, B.J.; Chun, J.S.; Kim, J.H. The role of selenium metabolism and selenoproteins in cartilage homeostasis and arthropathies. Exp. Mol. Med. 2020, 52, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; He, H.; Xiang, J.Q.; Yin, H.Q.; Hou, T. Selenium-Containing Proteins/Peptides from Plants: A Review on the Structures and Functions. J. Agric. Food Chem. 2020, 68, 15061–15073. [Google Scholar] [CrossRef] [PubMed]
- Flohé, L. Looking back at the early stages of redox biology. Antioxidants 2020, 9, 1254. [Google Scholar] [CrossRef]
- Orian, L.; Flohé, L. Selenium-Catalyzed Reduction of Hydroperoxides in Chemistry and Biology. Antioxidants 2021, 10, 1560. [Google Scholar] [CrossRef]
- Mills, G.C. Hemoglobin catabolism. I. Glutathione peroxidase, an erythrocyte enzyme which protects hemoglobin from oxidative breakdown. J. Biol. Chem. 1957, 229, 189. [Google Scholar]
- Flohé, L.; Günzler, W.A.; Schock, H.H. Glutathione peroxidase. Selenoenzyme. FEBS Lett. 1973, 32, 132. [Google Scholar] [CrossRef]
- Rotruck, J.T.; Pope, A.L.; Ganther, H.E.; Swanson, A.B.; Hafeman, D.G.; Hoekstra, W.G. Selenium—Biochemical Role as a Component of Glutathione Peroxidase. Science 1973, 179, 588–590. [Google Scholar] [CrossRef]
- Forstrom, J.W.; Zakowski, J.J.; Tappel, A.L. Identification of Catalytic Site of Rat-Liver Glutathione Peroxidase as Selenocysteine. Biochemistry 1978, 17, 2639–2644. [Google Scholar] [CrossRef] [PubMed]
- Epp, O.; Ladenstein, R.; Wendel, A. The refined structure of the selenoenzyme glutathione peroxidase at 0.2-nm resolution. Eur. J. Biochem. 1983, 133, 51–69. [Google Scholar] [CrossRef] [PubMed]
- Kraus, R.J.; Foster, S.J.; Ganther, H.E. Identification of selenocysteine in glutathione peroxidase by mass spectroscopy. Biochemistry 1983, 22, 5853. [Google Scholar] [CrossRef]
- Ursini, F.; Bindoli, A. The role of selenium peroxidases in the protection against oxidative damage of membranes. Chem. Phys. Lipids 1987, 44, 255–276. [Google Scholar] [CrossRef] [PubMed]
- Ursini, F.; Maiorino, M.; Brigelius-Flohé, R.; Aumann, K.D.; Roveri, A.; Schomburg, D.; Flohe, L. Diversity of glutathione peroxidases. Methods Enzymol. 1995, 252, 38–53. [Google Scholar]
- Mauri, P.; Benazzi, L.; Flohe, L.; Maiorino, M.; Pietta, P.G.; Pilawa, S.; Roveri, A.; Ursini, F. Versatility of selenium catalysis in PHGPx unraveled by LC/ESI-MS/MS. Biol. Chem. 2003, 384, 575–588. [Google Scholar] [CrossRef] [PubMed]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione Peroxidase-1 in Health and Disease: From Molecular Mechanisms to Therapeutic Opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef]
- Brigelius-Flohé, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta Gen. Subj. 2013, 1830, 3289–3303. [Google Scholar] [CrossRef]
- Orian, L.; Mauri, P.; Roveri, A.; Toppo, S.; Benazzi, L.; Bosello-Travain, V.; De Palma, A.; Maiorino, M.; Miotto, G.; Zaccarin, M.; et al. Selenocysteine oxidation in glutathione peroxidase catalysis: An MS-supported quantum mechanics study. Free Radic. Biol. Med. 2015, 87, 1–14. [Google Scholar] [CrossRef]
- Flohé, L.; Toppo, S.; Orian, L. The glutathione peroxidase family: Discoveries and mechanism. Free Radic. Biol. Med. 2022, 187, 113–122. [Google Scholar] [CrossRef]
- Berry, M.J.; Banu, L.; Larsen, P.R. Type-I Iodothyronine Deiodinase Is a Selenocysteine-Containing Enzyme. Nature 1991, 349, 438–440. [Google Scholar] [CrossRef] [PubMed]
- Köhrle, J. Local activation and inactivation of thyroid hormones: The deiodinase family. Mol. Cell. Endocrinol. 1999, 151, 103–119. [Google Scholar] [CrossRef]
- Bianco, A.C.; Salvatore, D.; Gereben, B.; Berry, M.J.; Larsen, P.R. Biochemistry, cellular and molecular biology, and physiological roles of the iodothyronine selenodeiodinases. Endocr. Rev. 2002, 23, 38–89. [Google Scholar] [CrossRef]
- Köhrle, J. Iodothyronine deiodinases. Methods Enzymol. 2002, 347, 125–167. [Google Scholar]
- Kuiper, G.G.J.M.; Kester, M.H.A.; Peeters, R.P.; Visser, T.J. Biochemical Mechanisms of Thyroid Hormone Deiodination. Thyroid 2005, 15, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.C.; Kim, B.W. Deiodinases: Implications of the local control of thyroid hormone action. J. Clin. Investig. 2006, 116, 2571–2579. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, U.; Schlicker, C.; Braun, D.; Koehrle, J.; Steegborn, C. Crystal structure of mammalian selenocysteine-dependent iodothyronine deiodinase suggests a peroxiredoxin-like catalytic mechanism. Proc. Natl. Acad. Sci. USA 2014, 111, 10526–10531. [Google Scholar] [CrossRef]
- Mondal, S.; Raja, K.; Schweizer, U.; Mugesh, G. Chemistry and Biology in the Biosynthesis and Action of Thyroid Hormones. Angew. Chem. Int. Ed. 2016, 55, 7606–7630. [Google Scholar] [CrossRef]
- Schweizer, U.; Towell, H.; Vit, A.; Rodriguez-Ruiz, A.; Steegborn, C. Structural aspects of thyroid hormone binding to proteins and competitive interactions with natural and synthetic compounds. Mol. Cell. Endocrinol. 2017, 458, 57–67. [Google Scholar] [CrossRef]
- van der Spek, A.H.; Fliers, E.; Boelen, A. The classic pathways of thyroid hormone metabolism. Mol. Cell. Endocrinol. 2017, 458, 29–38. [Google Scholar] [CrossRef]
- Bayse, C.A.; Marsan, E.S.; Garcia, J.R.; Tran-Thompson, A.T. Thyroxine binding to type III iodothyronine deiodinase. Sci. Rep. 2020, 10, 15401. [Google Scholar] [CrossRef] [PubMed]
- Steegborn, C.; Schweizer, U. Structure and Mechanism of Iodothyronine Deiodinases—What We Know, What We Don’t Know, and What Would Be Nice to Know. Exp. Clin. Endocrinol. Diabetes 2020, 128, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Ruiz, A.; Braun, D.; Pflug, S.; Brol, A.; Sylvester, M.; Steegborn, C.; Schweizer, U. Insights into the Mechanism of Human Deiodinase 1. Int. J. Mol. Sci. 2022, 23, 5361. [Google Scholar] [CrossRef]
- Köhrle, J.; Fradrich, C. Deiodinases control local cellular and systemic thyroid hormone availability. Free Radic. Biol. Med. 2022, 193, 59–79. [Google Scholar] [CrossRef] [PubMed]
- Arai, K.; Toba, H.; Yamamoto, N.; Ito, M.; Mikami, R. Modeling Type-1 Iodothyronine Deiodinase with Peptide-Based Aliphatic Diselenides: Potential Role of Highly Conserved His and Cys Residues as a General Acid Catalyst. Chem. Eur. J. 2023, 29, e202202387. [Google Scholar] [CrossRef]
- Reich, H.J.; Hoeger, C.A.; Willis, W.W., Jr. Organoselenium chemistry. A study of intermediates in the fragmentation of aliphatic keto selenoxides. Characterization of selenoxides, selenenamides and selenol seleninates by proton, carbon-13 and selenium-77 NMR. Tetrahedron 1985, 41, 4771. [Google Scholar] [CrossRef]
- Du Mont, W.-W.; Martens, A.; Pohl, S.; Saak, W. Reversible dismutation and coordination of bis(2,4,6-triisopropylphenyl) diselenide with iodine. A model study that relates to iodine intercalation between selenium chains. Inorg. Chem. 1990, 29, 4847–4848. [Google Scholar] [CrossRef]
- Martens-Von Salzen, A.; Meyer, H.U.; Du Mont, W.-W. Diselenides and iodine: Influence of solution equilibria between covalent compounds and charge-transfer complexes. Phosphorus Sulfur Silicon Relat. Elem. 1992, 67, 67–71. [Google Scholar] [CrossRef]
- Sano, T.; Masuda, R.; Sase, S.; Goto, K. Isolable small-molecule cysteine sulfenic acid. Chem. Commun. 2021, 57, 2479–2482. [Google Scholar] [CrossRef]
- Masuda, R.; Kimura, R.; Karasaki, T.; Sase, S.; Goto, K. Modeling the catalytic cycle of glutathione peroxidase by nuclear magnetic resonance spectroscopic analysis of selenocysteine selenenic acids. J. Am. Chem. Soc. 2021, 143, 6345–6350. [Google Scholar] [CrossRef]
- Masuda, R.; Kuwano, S.; Sase, S.; Bortoli, M.; Madabeni, A.; Orian, L.; Goto, K. Model study on the catalytic cycle of glutathione peroxidase utilizing selenocysteine-containing tripeptides: Elucidation of the protective bypass mechanism involving selenocysteine selenenic acids. Bull. Chem. Soc. Jpn. 2022, 95, 1360–1379. [Google Scholar] [CrossRef]
- Masuda, R.; Kuwano, S.; Goto, K. Modeling Selenoprotein Se-Nitrosation: Synthesis of a Se-Nitrososelenocysteine with Persistent Stability. J. Am. Chem. Soc. 2023, 145, 14184–14189. [Google Scholar] [CrossRef] [PubMed]
- Masuda, R.; Karasaki, T.; Sase, S.; Kuwano, S.; Goto, K. Highly Electrophilic Intermediates in the Bypass Mechanism of Glutathione Peroxidase: Synthesis, Reactivity, and Structures of Selenocysteine-Derived Cyclic Selenenyl Amides. Chem. Eur. J. 2023, 29, e202302615. [Google Scholar] [CrossRef] [PubMed]
- Masuda, R.; Goto, K. Modeling of selenocysteine-derived reactive intermediates utilizing a nano-sized molecular cavity as a protective cradle. Methods Enzymol. 2022, 662, 331–361. [Google Scholar]
- Sase, S.; Kakimoto, R.; Kimura, R.; Goto, K. Synthesis of a stable primary-alkyl-substituted selenenyl iodide and its hydrolytic conversion to the corresponding selenenic acid. Molecules 2015, 20, 21415–21420. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goto, K.; Kimura, R.; Masuda, R.; Karasaki, T.; Sase, S. Demonstration of the Formation of a Selenocysteine Selenenic Acid through Hydrolysis of a Selenocysteine Selenenyl Iodide Utilizing a Protective Molecular Cradle. Molecules 2023, 28, 7972. https://doi.org/10.3390/molecules28247972
Goto K, Kimura R, Masuda R, Karasaki T, Sase S. Demonstration of the Formation of a Selenocysteine Selenenic Acid through Hydrolysis of a Selenocysteine Selenenyl Iodide Utilizing a Protective Molecular Cradle. Molecules. 2023; 28(24):7972. https://doi.org/10.3390/molecules28247972
Chicago/Turabian StyleGoto, Kei, Ryutaro Kimura, Ryosuke Masuda, Takafumi Karasaki, and Shohei Sase. 2023. "Demonstration of the Formation of a Selenocysteine Selenenic Acid through Hydrolysis of a Selenocysteine Selenenyl Iodide Utilizing a Protective Molecular Cradle" Molecules 28, no. 24: 7972. https://doi.org/10.3390/molecules28247972
APA StyleGoto, K., Kimura, R., Masuda, R., Karasaki, T., & Sase, S. (2023). Demonstration of the Formation of a Selenocysteine Selenenic Acid through Hydrolysis of a Selenocysteine Selenenyl Iodide Utilizing a Protective Molecular Cradle. Molecules, 28(24), 7972. https://doi.org/10.3390/molecules28247972