
Drugging the Undruggable Trypanosoma brucei Monothiol Glutaredoxin 1

 , ,
, ,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
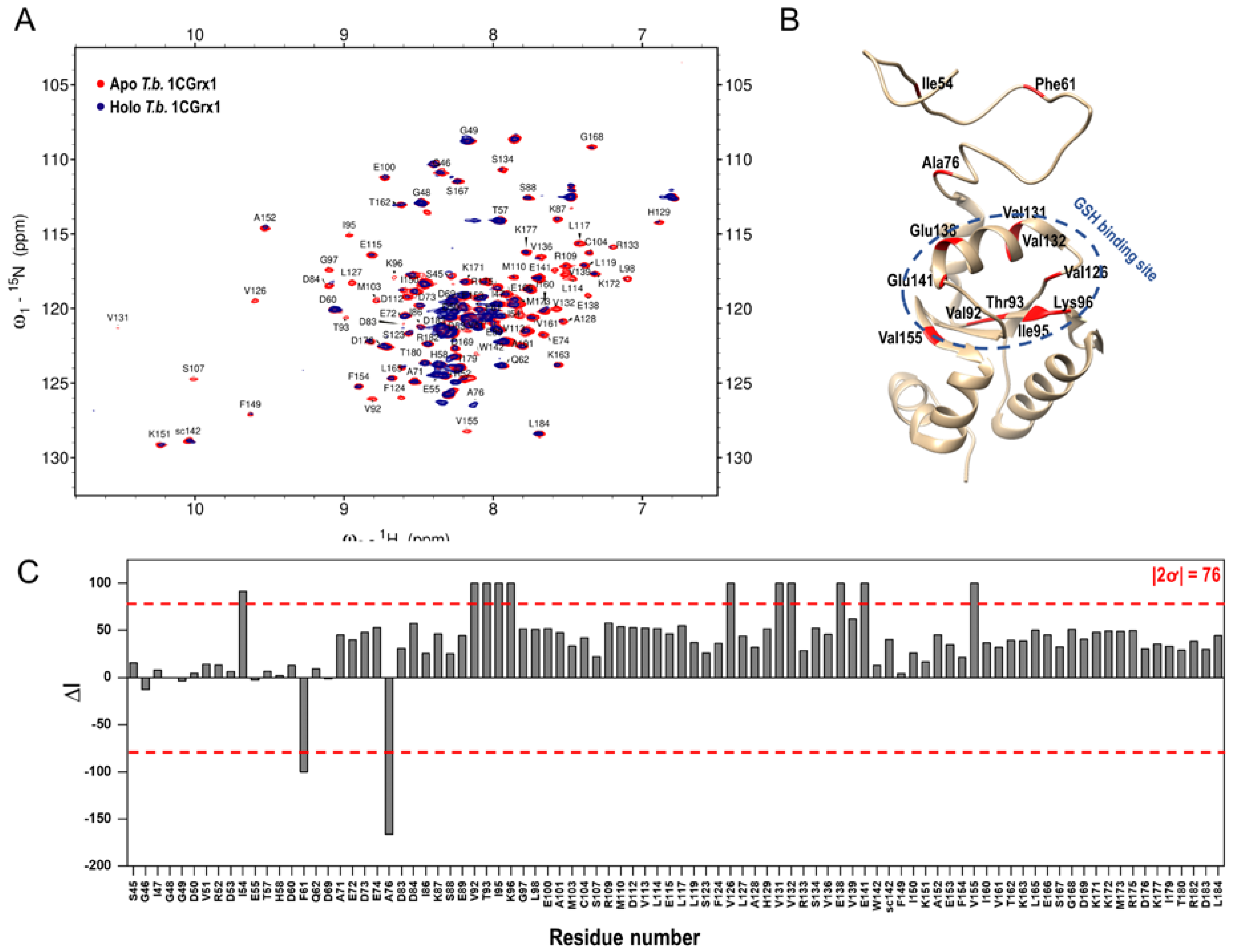
2.1. Fragment Library Screening by 1H-15N SOFAST-HMQC
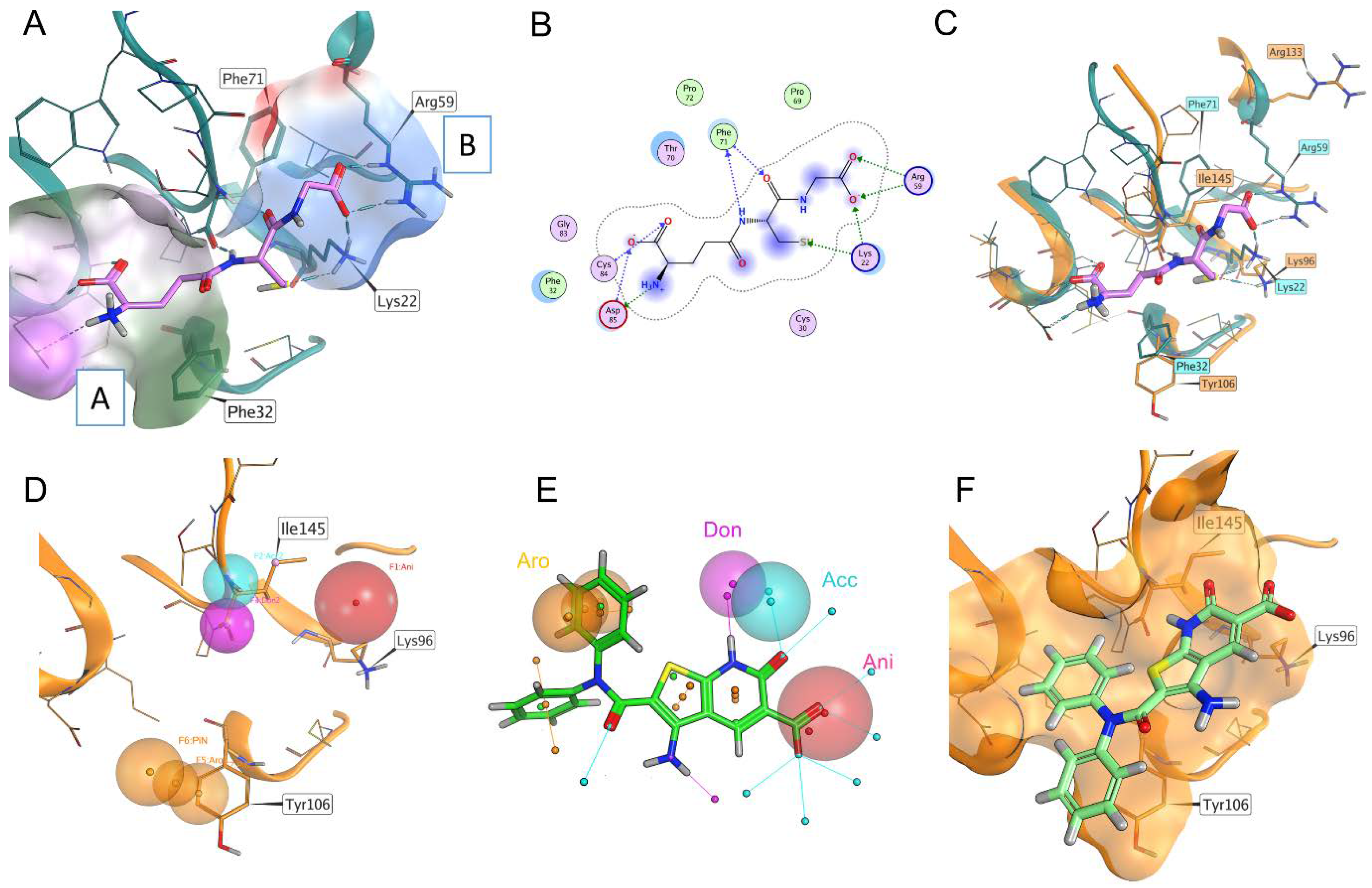
2.2. Structure-Based Virtual Screening Directed at the GSH/T(SH)2 Binding Site
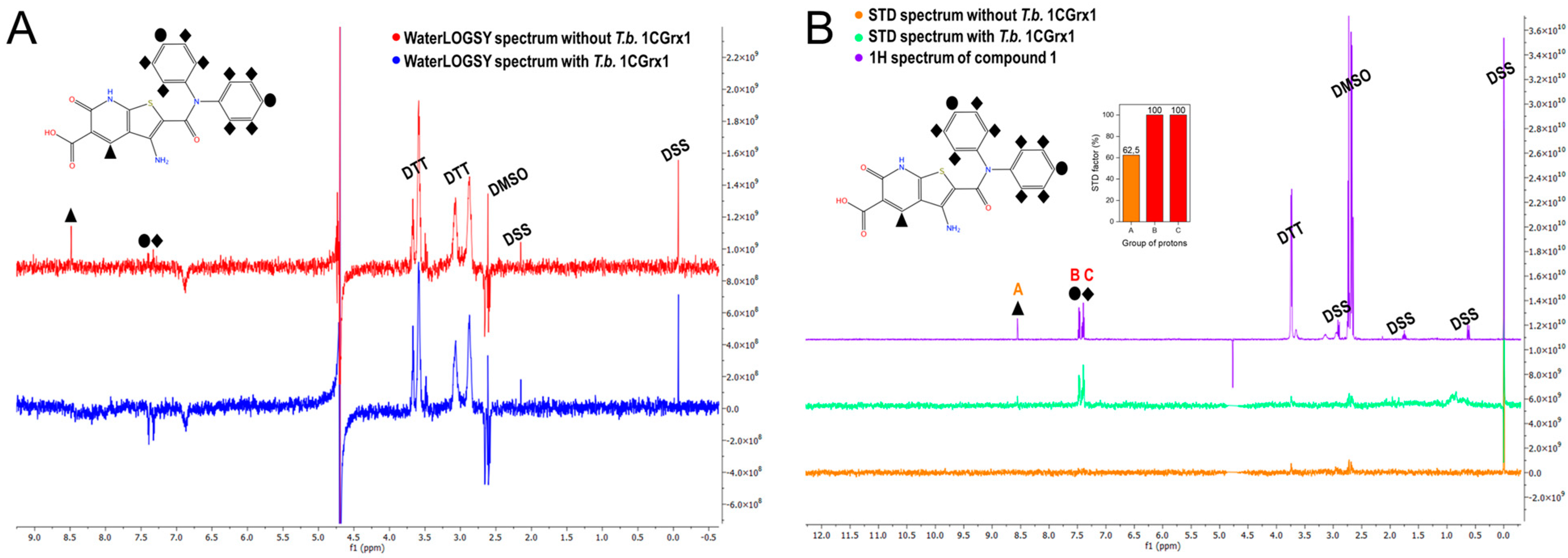
2.3. Biophysical Characterization of the Binding between T.b. 1CGrx1 and Hit Compounds by NMR
3. Materials and Methods
3.1. Computational Studies
3.1.1. Molecular Modeling
3.1.2. Pharmacophore Modeling and Molecular Docking
3.2. Recombinant T.b. 1CGrx1 Expression and Purification
3.3. Fragment Library Preparation and Characterization
3.4. NMR-Based Approaches
3.4.1. NMR-Based Fragment Screening
3.4.2. NMR Binding Analysis of the Virtual Screened Compounds
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Trypanosomiasis, Human African (Sleeping Sickness). Available online: https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness) (accessed on 27 November 2022).
- Rogers, D.J.; Williams, B.G. Monitoring trypanosomiasis in space and time. Parasitology 1993, 106, S77–S92. [Google Scholar] [CrossRef] [PubMed]
- Lutje, V.; Seixas, J.; Kennedy, A. Chemotherapy for second-stage human African trypanosomiasis. Cochrane Database Syst. Rev. 2013, 2013, CD006201. [Google Scholar] [CrossRef]
- Kande Betu Ku Mesu, V.; Mutombo Kalonji, W.; Bardonneau, C.; Valverde Mordt, O.; Ngolo Tete, D.; Blesson, S.; Simon, F.; Delhomme, S.; Bernhard, S.; Mahenzi Mbembo, H.; et al. Oral fexinidazole for stage 1 or early stage 2 African Trypanosoma brucei gambiense trypanosomiasis: A prospective, multicentre, open-label, cohort study. Lancet Glob. Health 2021, 9, e999–e1008. [Google Scholar] [CrossRef] [PubMed]
- Ilemobade, A.A. Tsetse and trypanosomosis in Africa: The challenges, the opportunities. Onderstepoort J. Vet. Res. 2009, 76, 35–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fairlamb, A.H.; Cerami, A. Metabolism and functions of trypanothione in the Kinetoplastida. Annu. Rev. Microbiol. 1992, 46, 695–729. [Google Scholar] [CrossRef]
- Manta, B.; Bonilla, M.; Fiestas, L.; Sturlese, M.; Salinas, G.; Bellanda, M.; Comini, M.A. Polyamine-Based Thiols in Trypanosomatids: Evolution, Protein Structural Adaptations, and Biological Functions. Antioxid. Redox Signal. 2018, 28, 463–486. [Google Scholar] [CrossRef]
- Lillig, C.H.; Berndt, C. Glutaredoxins in thiol/disulfide exchange. Antioxid. Redox Signal. 2013, 18, 1654–1665. [Google Scholar] [CrossRef]
- Ogata, F.T.; Branco, V.; Vale, F.F.; Coppo, L. Glutaredoxin: Discovery, redox defense and much more. Redox Biol. 2021, 43, 101975. [Google Scholar] [CrossRef]
- Comini, M.A.; Krauth-Siegel, R.L.; Bellanda, M. Mono- and dithiol glutaredoxins in the trypanothione-based redox metabolism of pathogenic trypanosomes. Antioxid. Redox Signal. 2013, 19, 708–722. [Google Scholar] [CrossRef] [Green Version]
- Filser, M.; Comini, M.A.; Molina-Navarro, M.M.; Dirdjaja, N.; Herrero, E.; Krauth-Siegel, R.L. Cloning, functional analysis, and mitochondrial localization of Trypanosoma brucei monothiol glutaredoxin-1. Biol. Chem. 2008, 389, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Comini, M.A.; Rettig, J.; Dirdjaja, N.; Hanschmann, E.-M.; Berndt, C.; Krauth-Siegel, R.L. Monothiol glutaredoxin-1 is an essential iron-sulfur protein in the mitochondrion of African trypanosomes. J. Biol. Chem. 2008, 283, 27785–27798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manta, B.; Pavan, C.; Sturlese, M.; Medeiros, A.; Crispo, M.; Berndt, C.; Krauth-Siegel, R.L.; Bellanda, M.; Comini, M.A. Iron-sulfur cluster binding by mitochondrial monothiol glutaredoxin-1 of Trypanosoma brucei: Molecular basis of iron-sulfur cluster coordination and relevance for parasite infectivity. Antioxid. Redox Signal. 2013, 19, 665–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturlese, M.; Manta, B.; Bertarello, A.; Bonilla, M.; Lelli, M.; Zambelli, B.; Grunberg, K.; Mammi, S.; Comini, M.A.; Bellanda, M. The lineage-specific, intrinsically disordered N-terminal extension of monothiol glutaredoxin 1 from trypanosomes contains a regulatory region. Sci. Rep. 2018, 8, 13716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balatti, G.E.; Barletta, G.P.; Parisi, G.; Tosatto, S.C.E.; Bellanda, M.; Fernandez-Alberti, S. Intrinsically Disordered Region Modulates Ligand Binding in Glutaredoxin 1 from Trypanosoma Brucei. J. Phys. Chem. B 2021, 125, 13366–13375. [Google Scholar] [CrossRef] [PubMed]
- Molina-Navarro, M.M.; Casas, C.; Piedrafita, L.; Bellí, G.; Herrero, E. Prokaryotic and eukaryotic monothiol glutaredoxins are able to perform the functions of Grx5 in the biogenesis of Fe/S clusters in yeast mitochondria. FEBS Lett. 2006, 580, 2273–2280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erlanson, D.A.; Davis, B.J.; Jahnke, W. Fragment-Based Drug Discovery: Advancing Fragments in the Absence of Crystal Structures. Cell Chem. Biol. 2019, 26, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, P.; Vendruscolo, M. Druggability of intrinsically disordered proteins. Adv. Exp. Med. Biol. 2015, 870, 383–400. [Google Scholar] [CrossRef]
- Mureddu, L.G.; Vuister, G.W. Fragment-Based Drug Discovery by NMR. Where Are the Successes and Where can It Be Improved? Front. Mol. Biosci. 2022, 9, 834453. [Google Scholar] [CrossRef]
- Iwema, T.; Picciocchi, A.; Traore, D.A.K.; Ferrer, J.-L.; Chauvat, F.; Jacquamet, L. Structural basis for delivery of the intact [Fe2S2] cluster by monothiol glutaredoxin. Biochemistry 2009, 48, 6041–6043. [Google Scholar] [CrossRef] [PubMed]
- OpenEye Scientific Software Inc OEChem v.2020. Available online: https://www.eyesopen.com/ (accessed on 4 December 2022).
- Molecular Networks GmbH CORINA.; Germany. Available online: https://mn-am.com/products/corina/ (accessed on 4 December 2022).
- Chemical Computing Group (CCG) Inc Molecular Operating Environment (MOE) v.2020.09. Available online: https://www.chemcomp.com/ (accessed on 4 December 2022).
- Baell, J.B. Observations on screening-based research and some concerning trends in the literature. Future Med. Chem. 2010, 2, 1529–1546. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [Green Version]
- Schanda, P.; Kupce, E.; Brutscher, B. SOFAST-HMQC experiments for recording two-dimensional heteronuclear correlation spectra of proteins within a few seconds. J. Biomol. NMR 2005, 33, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Tonelli, M.; Markley, J.L. NMRFAM-SPARKY: Enhanced software for biomolecular NMR spectroscopy. Bioinformatics 2015, 31, 1325–1327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williamson, M.P. Using chemical shift perturbation to characterise ligand binding. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 1–16. [Google Scholar] [CrossRef]
- Sturlese, M.; Lelli, M.; Manta, B.; Mammi, S.; Comini, M.A.; Bellanda, M. (1)H, (13)C and (15)N resonance assignment of the mature form of monothiol glutaredoxin 1 from the pathogen Trypanosoma brucei. Biomol. NMR Assign. 2015, 9, 143–146. [Google Scholar] [CrossRef]
- Schuffenhauer, A.; Ruedisser, S.; Marzinzik, A.L.; Jahnke, W.; Blommers, M.; Selzer, P.; Jacoby, E. Library design for fragment based screening. Curr. Top. Med. Chem. 2005, 5, 751–762. [Google Scholar] [CrossRef]
- Mayer, M.; Meyer, B. Characterization of Ligand Binding by Saturation Transfer Difference NMR Spectroscopy. Angew. Chem. Int. Ed. 1999, 38, 1784–1788. [Google Scholar] [CrossRef]
- Dalvit, C.; Fogliatto, G.; Stewart, A.; Veronesi, M.; Stockman, B. WaterLOGSY as a method for primary NMR screening: Practical aspects and range of applicability. J. Biomol. NMR 2001, 21, 349–359. [Google Scholar] [CrossRef]
- Hwang, T.L.; Shaka, A.J. Water Suppression That Works. Excitation Sculpting Using Arbitrary Wave-Forms and Pulsed-Field Gradients. J. Magn. Reson. Ser. A 1995, 112, 275–279. [Google Scholar] [CrossRef]
- Hajduk, P.J.; Huth, J.R.; Fesik, S.W. Druggability indices for protein targets derived from NMR-based screening data. J. Med. Chem. 2005, 48, 2518–2525. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Favaro, A.; Bolcato, G.; Comini, M.A.; Moro, S.; Bellanda, M.; Sturlese, M. Drugging the Undruggable Trypanosoma brucei Monothiol Glutaredoxin 1. Molecules 2023, 28, 1276. https://doi.org/10.3390/molecules28031276
Favaro A, Bolcato G, Comini MA, Moro S, Bellanda M, Sturlese M. Drugging the Undruggable Trypanosoma brucei Monothiol Glutaredoxin 1. Molecules. 2023; 28(3):1276. https://doi.org/10.3390/molecules28031276
Chicago/Turabian StyleFavaro, Annagiulia, Giovanni Bolcato, Marcelo A. Comini, Stefano Moro, Massimo Bellanda, and Mattia Sturlese. 2023. "Drugging the Undruggable Trypanosoma brucei Monothiol Glutaredoxin 1" Molecules 28, no. 3: 1276. https://doi.org/10.3390/molecules28031276
APA StyleFavaro, A., Bolcato, G., Comini, M. A., Moro, S., Bellanda, M., & Sturlese, M. (2023). Drugging the Undruggable Trypanosoma brucei Monothiol Glutaredoxin 1. Molecules, 28(3), 1276. https://doi.org/10.3390/molecules28031276