

Fluorinated Human Serum Albumin as Potential 19F Magnetic Resonance Imaging Probe


Abstract
:
1. Introduction
2. Results and Discussion
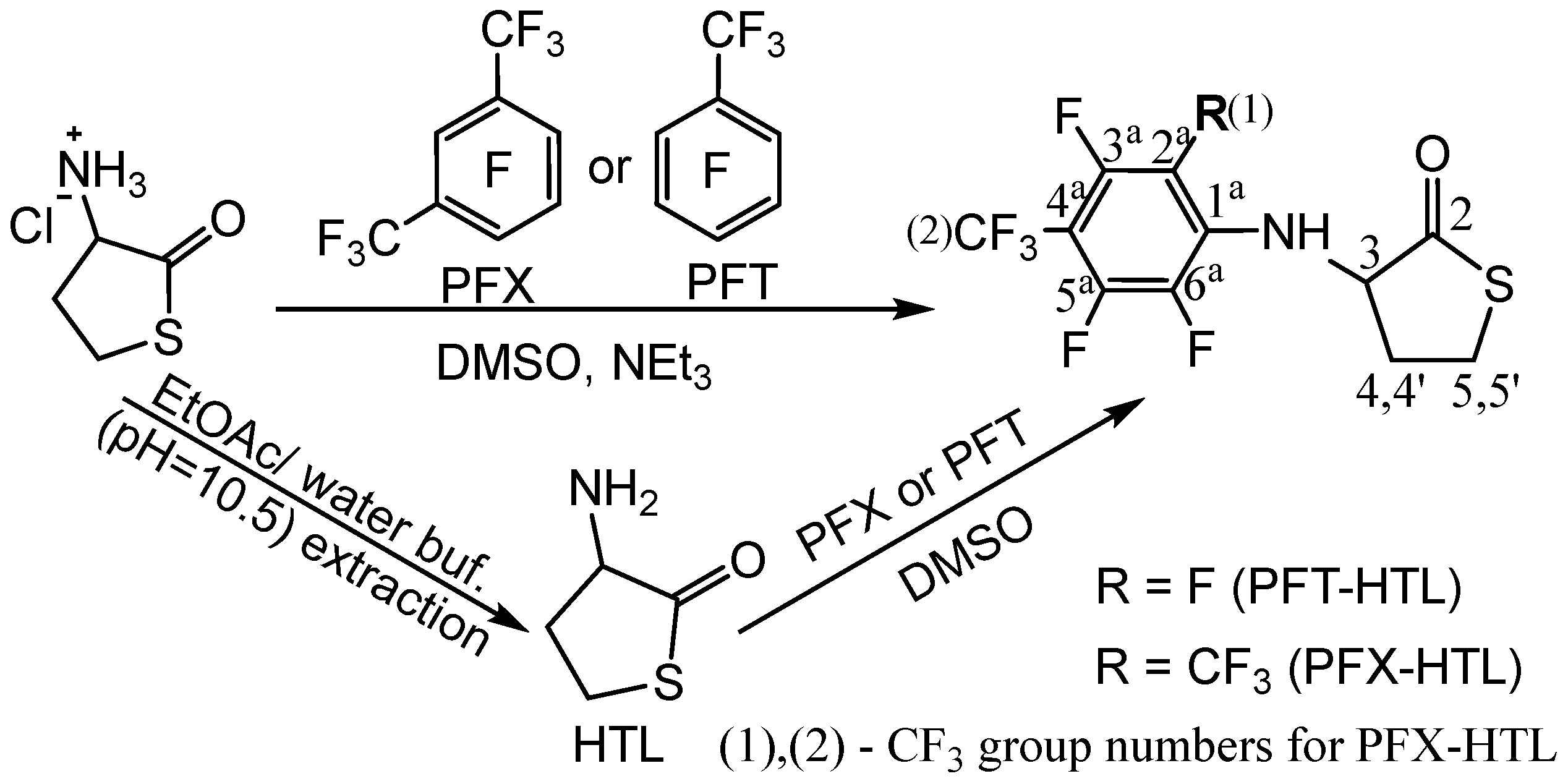
2.1. Synthesis of N-Fluorinated Homocysteine Thiolactone Derivatives
2.2. Synthesis and Characterization of Fluorinated HSA Conjugates
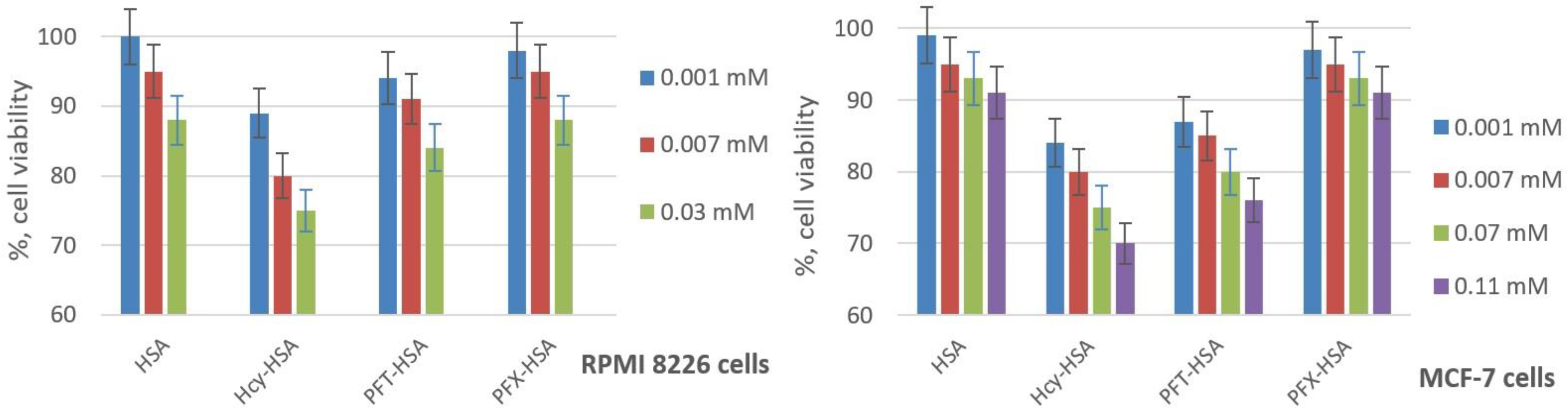
2.3. Cytotoxicity of HSA Conjugates
2.4. Dual-Labeled Albumin Conjugates Synthesis
2.5. Trypsin Degradation of Fluorinated HSA Conjugates
3. Materials and Methods
3.1. Chemicals
3.2. Physicochemical Characterization
3.3. Synthesis of S- and N-Fluorinated Derivatives of Homocysteine Thiolactone
3.3.1. Synthesis of Homocysteine Thiolactone (HTL) Free Base
3.3.2. Reaction of HTL Free Base with Perfluorotoluene (PFT) and Perfluoro-m-xylene (PFX) (The Method № 1)
3.3.3. Reaction of HTL Hydrochloride with Perfluorotoluene (PFT) and Perfluoro-m-xylene (PFX) in the Presence of a Triethylamine (The Method № 2)
3.3.4. N-(2,3,5,6-tetrafluoro-4-(trifluoromethyl)phenyl) Homocysteine Thiolactone (PFT-HTL)
3.3.5. N-(2,3,5-Trifluoro-4,6-bis(trifluoromethyl)phenyl) Homocysteine Thiolactone (PFX-HTL)
3.4. Synthesis of N-homocysteinylated Albumin (Hcy-HSA)
3.5. Synthesis of HSA-Cy5 and HSA-Cy7
3.6. Synthesis of Fluorinated Albumin Conjugates (PFT-HSA, PFX-HSA, PFT-HSA-Cy5, PFX-HSA-Cy5, PFT-HSA-Cy7, PFX-HSA-Cy7)
3.7. Synthesis of PFT-HSA Using the Mixture of S- and N-Substituted Derivatives of PFT
3.8. Susceptibility of Albumin Conjugates to Proteolysis
3.9. Cell Culture and Toxicity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Xiao, Y.-D.; Paudel, R.; Liu, J.; Ma, C.; Zhang, Z.-S.; Zhou, S.-K. MRI contrast agents: Classification and application (Review). Int. J. Mol. Med. 2016, 38, 1319–1326. [Google Scholar] [CrossRef]
- Angelovski, G. What We Can Really Do with Bioresponsive MRI Contrast Agents. Angew. Chem. Int. Ed. 2016, 55, 7038–7046. [Google Scholar] [CrossRef]
- Wahsner, J.; Gale, E.M.; Rodríguez-Rodríguez, A.; Caravan, P. Chemistry of MRI Contrast Agents: Current Challenges and New Frontiers. Chem. Rev. 2018, 119, 957–1057. [Google Scholar] [CrossRef]
- Geraldes, C.F.G.C.; Peters, J.A. MRI Contrast Agents in Glycobiology. Molecules 2022, 27, 8297. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Meade, T.J. Molecular Magnetic Resonance Imaging with Gd(III)-Based Contrast Agents: Challenges and Key Advances. J. Am. Chem. Soc. 2019, 141, 17025–17041. [Google Scholar] [CrossRef]
- Kim, H.-K.; Lee, G.H.; Chang, Y. Gadolinium as an MRI contrast agent. Future Med. Chem. 2018, 10, 639–661. [Google Scholar] [CrossRef]
- Caspani, S.; Magalhães, R.; Araújo, J.P.; Sousa, C.T. Magnetic Nanomaterials as Contrast Agents for MRI. Materials 2020, 13, 2586. [Google Scholar] [CrossRef]
- Petrov, K.D.; Chubarov, A.S. Magnetite Nanoparticles for Biomedical Applications. Encyclopedia 2022, 2, 1811–1828. [Google Scholar] [CrossRef]
- Popova, V.; Dmitrienko, E.; Chubarov, A. Magnetic Nanocomposites and Imprinted Polymers for Biomedical Applications of Nucleic Acids. Magnetochemistry 2023, 9, 12. [Google Scholar] [CrossRef]
- Chubarov, A.S. Serum Albumin for Magnetic Nanoparticles Coating. Magnetochemistry 2022, 8, 13. [Google Scholar] [CrossRef]
- Thomsen, H.S.; Morcos, S.K.; Almén, T.; Bellin, M.-F.; Bertolotto, M.; Bongartz, G.; Clement, O.; Leander, P.; Heinz-Peer, G.; Reimer, P.; et al. Nephrogenic systemic fibrosis and gadolinium-based contrast media: Updated ESUR Contrast Medium Safety Committee guidelines. Eur. Radiol. 2013, 23, 307–318. [Google Scholar] [CrossRef]
- Pasquini, L.; Napolitano, A.; Visconti, E.; Longo, D.; Romano, A.; Tomà, P.; Espagnet, M.C.R. Gadolinium-Based Contrast Agent-Related Toxicities. CNS Drugs 2018, 32, 229–240. [Google Scholar] [CrossRef]
- Ramalho, J.; Ramalho, M. Gadolinium Deposition and Chronic Toxicity. Magn. Reson. Imaging Clin. N. Am. 2017, 25, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Malikova, H.; Holesta, M. Gadolinium Contrast Agents—Are they Really Safe? J. Vasc. Access 2017, 18 (Suppl. S2), S1–S7. [Google Scholar] [CrossRef] [PubMed]
- Akakuru, O.U.; Iqbal, M.Z.; Saeed, M.; Liu, C.; Paunesku, T.; Woloschak, G.; Hosmane, N.S.; Wu, A. The Transition from Metal-Based to Metal-Free Contrast Agents for T1 Magnetic Resonance Imaging Enhancement. Bioconjug. Chem. 2019, 30, 2264–2286. [Google Scholar] [CrossRef]
- Dobrynin, S.; Kutseikin, S.; Morozov, D.; Krumkacheva, O.; Spitsyna, A.; Gatilov, Y.; Silnikov, V.; Angelovski, G.; Bowman, M.K.; Kirilyuk, I.; et al. Human Serum Albumin Labelled with Sterically-Hindered Nitroxides as Potential MRI Contrast Agents. Molecules 2020, 25, 1709. [Google Scholar] [CrossRef]
- Nguyen, H.V.-T.; Detappe, A.; Harvey, P.; Gallagher, N.; Mathieu, C.; Agius, M.P.; Zavidij, O.; Wang, W.; Jiang, Y.; Rajca, A.; et al. Pro-organic radical contrast agents (“pro-ORCAs”) for real-time MRI of pro-drug activation in biological systems. Polym. Chem. 2020, 11, 4768–4779. [Google Scholar] [CrossRef]
- Nguyen, H.V.-T.; Detappe, A.; Gallagher, N.M.; Zhang, H.; Harvey, P.; Yan, C.; Mathieu, C.; Golder, M.R.; Jiang, Y.; Ottaviani, M.F.; et al. Triply Loaded Nitroxide Brush-Arm Star Polymers Enable Metal-Free Millimetric Tumor Detection by Magnetic Resonance Imaging. ACS Nano 2018, 12, 11343–11354. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Lloveras, V.; Lope-Piedrafita, S.; Calero-Pérez, P.; Wu, S.; Candiota, A.P.; Vidal-Gancedo, J. Metal-Free Radical Dendrimers as MRI Contrast Agents for Glioblastoma Diagnosis: Ex Vivo and In Vivo Approaches. Biomacromolecules 2022, 23, 2767–2777. [Google Scholar] [CrossRef]
- Bartusik-Aebisher, D.; Bober, Z.; Zalejska-Fiolka, J.; Kawczyk-Krupka, A.; Aebisher, D. Multinuclear MRI in Drug Discovery. Molecules 2022, 27, 6493. [Google Scholar] [CrossRef]
- Godovikova, T.S.; Lisitskiy, V.A.; Antonova, N.M.; Popova, T.V.; Zakharova, O.D.; Chubarov, A.S.; Koptyug, I.V.; Sagdeev, R.Z.; Kaptein, R.; Akulov, A.E.; et al. Ligand-Directed Acid-Sensitive Amidophosphate 5-Trifluoromethyl-2′-Deoxyuridine Conjugate as a Potential Theranostic Agent. Bioconjug. Chem. 2013, 24, 780–795. [Google Scholar] [CrossRef] [PubMed]
- Dawid, J.; Krawczyk, T. F-19 MRI Probes for Multimodal Imaging. Chem. A Eur. J. 2022, 28, e202102556. [Google Scholar] [CrossRef]
- Chen, H.; Viel, S.; Ziarelli, F.; Peng, L. 19F NMR: A valuable tool for studying biological events. Chem. Soc. Rev. 2013, 42, 7971–7982. [Google Scholar] [CrossRef] [PubMed]
- Dahanayake, J.N.; Kasireddy, C.; Karnes, J.P.; Verma, R.; Steinert, R.M.; Hildebrandt, D.; Hull, O.A.; Ellis, J.M.; Mitchell-Koch, K.R. Progress in Our Understanding of 19F Chemical Shifts. In Annual Reports on NMR Spectroscopy, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2018; Volume 93, pp. 281–365. [Google Scholar]
- Ruiz-Cabello, J.; Barnett, B.P.; Bottomley, P.A.; Bulte, J.W. Fluorine (19F) MRS and MRI in biomedicine. NMR Biomed. 2011, 24, 114–129. [Google Scholar] [CrossRef]
- Cho, M.H.; Shin, S.H.; Park, S.H.; Kadayakkara, D.K.; Kim, D.; Choi, Y. Targeted, Stimuli-Responsive, and Theranostic 19F Magnetic Resonance Imaging Probes. Bioconjug. Chem. 2019, 30, 2502–2518. [Google Scholar] [CrossRef] [PubMed]
- Tirotta, I.; Dichiarante, V.; Pigliacelli, C.; Cavallo, G.; Terraneo, G.; Bombelli, F.B.; Metrangolo, P.; Resnati, G. 19F Magnetic Resonance Imaging (MRI): From Design of Materials to Clinical Applications. Chem. Rev. 2015, 115, 1106–1129. [Google Scholar] [CrossRef]
- Cobb, S.L.; Murphy, C.D. 19F NMR applications in chemical biology. J. Fluor. Chem. 2009, 130, 132–143. [Google Scholar] [CrossRef]
- Qin, L.; Sheridan, C.; Gao, J. Synthesis of tetrafluorinated aromatic amino acids with distinct signatures in 19F NMR. Org. Lett. 2012, 14, 528–531. [Google Scholar] [CrossRef]
- Zhang, C.; Yan, K.; Fu, C.; Peng, H.; Hawker, C.J.; Whittaker, A.K. Biological Utility of Fluorinated Compounds: From Materials Design to Molecular Imaging, Therapeutics and Environmental Remediation. Chem. Rev. 2021, 122, 167–208. [Google Scholar] [CrossRef]
- Meyer, A.; Dechert, S.; Dey, S.; Höbartner, C.; Bennati, M. Measurement of Angstrom to Nanometer Molecular Distances with 19F Nuclear Spins by EPR/ENDOR Spectroscopy. Angew. Chem. Int. Ed. 2020, 59, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Kehl, A.; Hiller, M.; Hecker, F.; Tkach, I.; Dechert, S.; Bennati, M.; Meyer, A. Resolution of chemical shift anisotropy in 19F ENDOR spectroscopy at 263 GHz/9.4 T. J. Magn. Reson. 2021, 333, 107091. [Google Scholar] [CrossRef]
- Asanbaeva, N.B.; Sukhanov, A.A.; Diveikina, A.A.; Rogozhnikova, O.Y.; Trukhin, D.V.; Tormyshev, V.M.; Chubarov, A.S.; Maryasov, A.G.; Genaev, A.M.; Shernyukov, A.V.; et al. Application of W-band 19F electron nuclear double resonance (ENDOR) spectroscopy to distance measurement using a trityl spin probe and a fluorine label. Phys. Chem. Chem. Phys. 2022, 24, 5982–6001. [Google Scholar] [CrossRef]
- Wu, T.; Li, A.; Chen, K.; Peng, X.; Zhang, J.; Jiang, M.; Chen, S.; Zheng, X.; Zhou, X.; Jiang, Z.-X. Perfluoro-tert-butanol: A cornerstone for high performance fluorine-19 magnetic resonance imaging. Chem. Commun. 2021, 57, 7743–7757. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cui, J.; Li, C.; Zhou, H.; Chang, J.; Aras, O.; An, F. 19F MRI Nanotheranostics for Cancer Management: Progress and Prospects. Chemmedchem 2022, 17, e202100701. [Google Scholar] [CrossRef] [PubMed]
- Mali, A.; Kaijzel, E.L.; Lamb, H.J.; Cruz, L.J. 19F-nanoparticles: Platform for in vivo delivery of fluorinated biomaterials for 19F-MRI. J. Control. Release 2021, 338, 870–889. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.M.; Gigliobianco, M.R.; Firouzabadi, B.M.; Censi, R.; Di Martino, P. Nanotechnology as a Versatile Tool for 19F-MRI Agent’s Formulation: A Glimpse into the Use of Perfluorinated and Fluorinated Compounds in Nanoparticles. Pharmaceutics 2022, 14, 382. [Google Scholar] [CrossRef]
- Wu, L.; Liu, F.; Liu, S.; Xu, X.; Liu, Z.; Sun, X. Perfluorocarbons-Based 19F Magnetic Resonance Imaging in Biomedicine. Int. J. Nanomed. 2020, 15, 7377–7395. [Google Scholar] [CrossRef]
- Zhu, L.; Li, Y.; Jiang, M.; Ke, C.; Long, H.; Qiu, M.; Zhang, L.; Ye, C.; Zhou, X.; Jiang, Z.-X.; et al. Self-Assembly of Precisely Fluorinated Albumin for Dual Imaging-Guided Synergistic Chemo–Photothermal–Photodynamic Cancer Therapy. ACS Appl. Mater. Interfaces 2023, 15, 2665–2678. [Google Scholar] [CrossRef]
- Yue, X.; Feng, Y.; Yu, Y.B. Synthesis and characterization of fluorinated conjugates of albumin. J. Fluor. Chem. 2013, 152, 173–181. [Google Scholar] [CrossRef]
- Yeo, S.K.; Shepelytskyi, Y.; Grynko, V.; Albert, M.S. Molecular Imaging of Fluorinated Probes for Tau Protein and Amyloid-β Detection. Molecules 2020, 25, 3413. [Google Scholar] [CrossRef]
- Yanagisawa, D.; Ibrahim, N.F.; Taguchi, H.; Morikawa, S.; Tomiyama, T.; Tooyama, I. Fluorine-19 Magnetic Resonance Imaging for Detection of Amyloid β Oligomers Using a Keto Form of Curcumin Derivative in a Mouse Model of Alzheimer’s Disease. Molecules 2021, 26, 1362. [Google Scholar] [CrossRef] [PubMed]
- Tooyama, I.; Yanagisawa, D.; Taguchi, H.; Kato, T.; Hirao, K.; Shirai, N.; Sogabe, T.; Ibrahim, N.F.; Inubushi, T.; Morikawa, S. Amyloid imaging using fluorine-19 magnetic resonance imaging (19F-MRI). Ageing Res. Rev. 2016, 30, 85–94. [Google Scholar] [CrossRef]
- Modo, M. 19F Magnetic Resonance Imaging and Spectroscopy in Neuroscience. Neuroscience 2021, 474, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Bouvain, P.; Temme, S.; Flögel, U. Hot spot 19F magnetic resonance imaging of inflammation. WIREs Nanomed. Nanobiotechnol. 2020, 12, e1639. [Google Scholar] [CrossRef]
- De Simone, G.; di Masi, A.; Ascenzi, P. Serum Albumin: A Multifaced Enzyme. Int. J. Mol. Sci. 2021, 22, 10086. [Google Scholar] [CrossRef]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Asp. Med. 2012, 33, 209–290. [Google Scholar] [CrossRef] [PubMed]
- Merlot, A.M.; Kalinowski, D.S.; Richardson, D.R. Unraveling the mysteries of serum albumin—More than just a serum protein. Front. Physiol. 2014, 5, 299. [Google Scholar] [CrossRef]
- Belinskaia, D.A.; Voronina, P.A.; Shmurak, V.I.; Jenkins, R.O.; Goncharov, N.V. Serum Albumin in Health and Disease: Esterase, Antioxidant, Transporting and Signaling Properties. Int. J. Mol. Sci. 2021, 22, 10318. [Google Scholar] [CrossRef]
- Nishi, K.; Yamasaki, K.; Otagiri, M. Serum Albumin, Lipid and Drug Binding. Vertebr. Invertebr. Respir. Proteins Lipoproteins Other Body Fluid Proteins 2020, 94, 383–397. [Google Scholar]
- van der Vusse, G.J. Albumin as Fatty Acid Transporter. Drug Metab. Pharmacokinet. 2009, 24, 300–307. [Google Scholar] [CrossRef]
- Otagiri, M.; Giam Chuang, V.T. Albumin in Medicine: Pathological and Clinical Applications; Springer: Singapore, 2016; pp. 1–277. [Google Scholar] [CrossRef]
- Kratz, F. Albumin as a drug carrier: Design of prodrugs, drug conjugates and nanoparticles. J. Control. Release 2008, 132, 171–183. [Google Scholar] [CrossRef] [PubMed]
- Kragh-Hansen, U. Human Serum Albumin: A Multifunctional Protein. In Albumin in Medicine: Pathological and Clinical Applications; Springer: Singapore, 2016; pp. 1–24. [Google Scholar]
- Sockolosky, J.T.; Szoka, F.C. The neonatal Fc receptor, FcRn, as a target for drug delivery and therapy. Adv. Drug Deliv. Rev. 2015, 91, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Sleep, D.; Cameron, J.; Evans, L.R. Albumin as a versatile platform for drug half-life extension. Biochim. Biophys. Acta BBA Gen. Subj. 2013, 1830, 5526–5534. [Google Scholar] [CrossRef]
- Larsen, M.T.; Kuhlmann, M.; Hvam, M.L.; Howard, K.A. Albumin-based drug delivery: Harnessing nature to cure disease. Mol. Cell. Ther. 2016, 4, 3. [Google Scholar] [CrossRef]
- Chubarov, A.S.; Shakirov, M.M.; Koptyug, I.V.; Sagdeev, R.Z.; Knorre, D.G.; Godovikova, T.S. Synthesis and characterization of fluorinated homocysteine derivatives as potential molecular probes for 19F magnetic resonance spectroscopy and imaging. Bioorg. Med. Chem. Lett. 2011, 21, 4050–4053. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, X. Simple bioconjugate chemistry serves great clinical advances: Albumin as a versatile platform for diagnosis and precision therapy. Chem. Soc. Rev. 2016, 45, 1432–1456. [Google Scholar] [CrossRef] [PubMed]
- Bern, M.; Sand, K.M.K.; Nilsen, J.; Sandlie, I.; Andersen, J.T. The role of albumin receptors in regulation of albumin homeostasis: Implications for drug delivery. J. Control. Release 2015, 211, 144–162. [Google Scholar] [CrossRef]
- Sikder, S.; Gote, V.; Alshamrani, M.; Sicotte, J.; Pal, D. Long-term delivery of protein and peptide therapeutics for cancer therapies. Expert Opin. Drug Deliv. 2019, 16, 1113–1131. [Google Scholar] [CrossRef]
- Popova, T.V.; Khan, H.; Chubarov, A.S.; Lisitskiy, V.A.; Antonova, N.M.; Akulov, A.E.; Shevelev, O.B.; Zavjalov, E.L.; Silnikov, V.N.; Ahmad, S.; et al. Biotin-decorated anti-cancer nucleotide theranostic conjugate of human serum albumin: Where the seed meets the soil? Bioorg. Med. Chem. Lett. 2018, 28, 260–264. [Google Scholar] [CrossRef]
- Lisitskiy, V.A.; Khan, H.; Popova, T.V.; Chubarov, A.S.; Zakharova, O.D.; Akulov, A.E.; Shevelev, O.B.; Zavjalov, E.L.; Koptyug, I.V.; Moshkin, M.P.; et al. Multifunctional human serum albumin-therapeutic nucleotide conjugate with redox and pH-sensitive drug release mechanism for cancer theranostics. Bioorg. Med. Chem. Lett. 2017, 27, 3925–3930. [Google Scholar] [CrossRef]
- Chubarov, A.S.; Zakharova, O.D.; Koval, O.A.; Romaschenko, A.V.; Akulov, A.E.; Zavjalov, E.L.; Razumov, I.A.; Koptyug, I.V.; Knorre, D.G.; Godovikova, T.S. Design of protein homocystamides with enhanced tumor uptake properties for 19F magnetic resonance imaging. Bioorg. Med. Chem. 2015, 23, 6943–6954. [Google Scholar] [CrossRef]
- Chubarov, A.S. Homocysteine Thiolactone: Biology and Chemistry. Encyclopedia 2021, 1, 445–459. [Google Scholar] [CrossRef]
- Präbst, K.; Engelhardt, H.; Ringgeler, S.; Hübner, H. Basic Colorimetric Proliferation Assays: MTT, WST, and Resazurin. In Cell Viability Assays; Gilbert, D.F., Friedrich, O., Eds.; Springer: New York, NY, USA, 2017; Volume 1601, pp. 1–17. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Espeel, P.; Du Prez, F.E. One-pot multi-step reactions based on thiolactone chemistry: A powerful synthetic tool in polymer science. Eur. Polym. J. 2015, 62, 247–272. [Google Scholar] [CrossRef]
- Espeel, P.; Prez, F.E. One-Pot Double Modification of Polymers Based on Thiolactone Chemistry. Adv. Polym. Sci. 2015, 269, 105–132. [Google Scholar] [CrossRef]
- Frank, D.; Espeel, P.; Claessens, S.; Mes, E.; Du Prez, F.E. Synthesis of thiolactone building blocks as potential precursors for sustainable functional materials. Tetrahedron 2016, 72, 6616–6625. [Google Scholar] [CrossRef]
- Cui, H.-L. Recent Advances in DMSO-Based Direct Synthesis of Heterocycles. Molecules 2022, 27, 8480. [Google Scholar] [CrossRef]
- Bisconti, M.; Grosjean, P.; Arcolia, V.; Simon, J.-F.; Hennebert, E. Influence of Two Widely Used Solvents, Ethanol and Dimethyl Sulfoxide, on Human Sperm Parameters. Int. J. Mol. Sci. 2022, 24, 505. [Google Scholar] [CrossRef]
- Kuroda, K.; Komori, T.; Ishibashi, K.; Uto, T.; Kobayashi, I.; Kadokawa, R.; Kato, Y.; Ninomiya, K.; Takahashi, K.; Hirata, E. Non-aqueous, zwitterionic solvent as an alternative for dimethyl sulfoxide in the life sciences. Commun. Chem. 2020, 3, 163. [Google Scholar] [CrossRef]
- Vigneaud, V.; Patterson, W.I.; Hunt, M. Opeining of the Ring of the Thiolactone of Homocysteine. J. Biol. Chem 1938, 126, 217–231. [Google Scholar] [CrossRef]
- Martins, M.B.; Carvalho, I. Diketopiperazines: Biological activity and synthesis. Tetrahedron 2007, 63, 9923–9932. [Google Scholar] [CrossRef]
- Jakubowski, H. Mechanism of the Condensation of Homocysteine Thiolactone with Aldehydes. Chem. A Eur. J. 2006, 12, 8039–8043. [Google Scholar] [CrossRef]
- Garel, J.; Tawfik, D.S. Mechanism of Hydrolysis and Aminolysis of Homocysteine Thiolactone. Chem. A Eur. J. 2006, 12, 4144–4152. [Google Scholar] [CrossRef] [PubMed]
- Kwan, E.E.; Zeng, Y.; Besser, H.A.; Jacobsen, E.N. Concerted nucleophilic aromatic substitutions. Nat. Chem. 2018, 10, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Chambers, R.D.; Martin, P.A.; Sandford, G.; Williams, D.L.H. Mechanisms of reactions of halogenated compounds: Part 7. Effects of fluorine and other groups as substituents on nucleophilic aromatic substitution. J. Fluor. Chem. 2008, 129, 998–1002. [Google Scholar] [CrossRef]
- Muir, M.; Baker, J. A simple calculational model for predicting the site for nucleophilic substitution in aromatic perfluorocarbons. J. Fluor. Chem. 2005, 126, 727–738. [Google Scholar] [CrossRef]
- Kovtonyuk, V.N.; Gatilov, Y.V.; Nikul’Shin, P.V.; Bredikhin, R.A. Synthesis of Polyfluorinated Thia- and Oxathiacalixarenes Based on Perfluoro-m-xylene. Molecules 2021, 26, 526. [Google Scholar] [CrossRef] [PubMed]
- Mehta, V.D.; Kulkarni, P.V.; Mason, R.P.; Constantinescu, A.; Antich, P.P. Fluorinated Proteins as Potential 19F Magnetic Resonance Imaging and Spectroscopy Agents. Bioconjug. Chem. 1994, 5, 257–261. [Google Scholar] [CrossRef]
- Chubarov, A.; Spitsyna, A.; Krumkacheva, O.; Mitin, D.; Suvorov, D.; Tormyshev, V.; Fedin, M.; Bowman, M.K.; Bagryanskaya, E. Reversible Dimerization of Human Serum Albumin. Molecules 2021, 26, 108. [Google Scholar] [CrossRef]
- Jakubowski, H. Homocysteine Modification in Protein Structure/Function and Human Disease. Physiol. Rev. 2019, 99, 555–604. [Google Scholar] [CrossRef]
- Jakubowski, H. Molecular basis of homocysteine toxicity in humans. Cell. Mol. Life Sci. 2004, 61, 470–487. [Google Scholar] [CrossRef] [PubMed]
- Chi, C.; Du, Y.; Ye, J.; Kou, D.; Qiu, J.; Wang, J.; Tian, J.; Chen, X. Intraoperative Imaging-Guided Cancer Surgery: From Current Fluorescence Molecular Imaging Methods to Future Multi-Modality Imaging Technology. Theranostics 2014, 4, 1072–1084. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Xu, A.; Yu, Y.; Fu, C.; Liang, G. Biomedical Applications of Fluorescent and Magnetic Resonance Imaging Dual-Modality Probes. Chembiochem 2019, 20, 499–510. [Google Scholar] [CrossRef]
- Głowacki, R.; Jakubowski, H. Cross-talk between Cys34 and Lysine Residues in Human Serum Albumin Revealed by N-Homocysteinylation. J. Biol. Chem. 2004, 279, 10864–10871. [Google Scholar] [CrossRef]
- McIntyre, J.O.; Matrisian, L.M. Molecular imaging of proteolytic activity in cancer. J. Cell. Biochem. 2003, 90, 1087–1097. [Google Scholar] [CrossRef]
- Mellet, P.; Massot, P.; Madelin, G.; Marque, S.R.A.; Harté, E.; Franconi, J.-M.; Thiaudière, E. New Concepts in Molecular Imaging: Non-Invasive MRI Spotting of Proteolysis Using an Overhauser Effect Switch. PLoS ONE 2009, 4, e5244. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Hong, H.; Zhang, Y.; Cai, W. Molecular Imaging of Proteases in Cancer. Cancer Growth Metastasis 2009, 2, CGM.S2814. [Google Scholar] [CrossRef]
- Mahmood, U.; Weissleder, R. Near-infrared optical imaging of proteases in cancer. Mol. Cancer Ther. 2003, 2, 489–496. [Google Scholar]
- Koonjoo, N.; Parzy, E.; Massot, P.; Lepetit-Coiffé, M.; Marque, S.R.A.; Franconi, J.-M.; Thiaudiere, E.; Mellet, P. In vivo Overhauser-enhanced MRI of proteolytic activity. Contrast Media Mol. Imaging 2014, 9, 363–371. [Google Scholar] [CrossRef]
- Janatova, J.; Fuller, J.K.; Hunter, M.J. The Heterogeneity of Bovine Albumin with Respect to Sulfhydryl and Dimer Content. J. Biol. Chem. 1968, 243, 3612–3622. [Google Scholar] [CrossRef]
- Perczel, A.; Hollósi, M.; Tusn’dy, G.; Fasman, G.D. Convex constraint analysis: A natural deconvolution of circular dichroism curves of proteins. Protein Eng. Des. Sel. 1991, 4, 669–679. [Google Scholar] [CrossRef]
- Strohalm, M.; Kavan, D.; Novák, P.; Volný, M.; Havlíček, V. mMass 3: A Cross-Platform Software Environment for Precise Analysis of Mass Spectrometric Data. Anal. Chem. 2010, 82, 4648–4651. [Google Scholar] [CrossRef] [PubMed]
- Niedermeyer, T.H.J.; Strohalm, M. mMass as a Software Tool for the Annotation of Cyclic Peptide Tandem Mass Spectra. PLoS ONE 2012, 7, e44913. [Google Scholar] [CrossRef] [PubMed]
- Jackson, P.; Attalla, M.I. N-Nitrosopiperazines Form at High PH in Post-Combustion Capture Solutions Containing Piperazine: A Low-Energy Collisional Behaviour Study. Rapid Commun. Mass Spectrom. 2010, 24, 3567–3577. [Google Scholar] [CrossRef]
- Tormyshev, V.M.; Chubarov, A.S.; Krumkacheva, O.A.; Trukhin, D.V.; Rogozhnikova, O.Y.; Spitsyna, A.S.; Kuzhelev, A.A.; Koval, V.; Fedin, M.V.; Godovikova, T.S.; et al. Methanethiosulfonate Derivative of OX063 Trityl: A Promising and Efficient Reagent for Side-Directed Spin Labeling of Proteins. Chem. A Eur. J. 2020, 26, 2705–2712. [Google Scholar] [CrossRef] [PubMed]
- Ishima, Y.; Maruyama, T.; Otagiri, M.; Chuang, V.T.G.; Ishida, T. The New Delivery Strategy of Albumin Carrier Utilizing the Interaction with Albumin Receptors. Chem. Pharm. Bull. 2022, 70, 330–333. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Synthesis Step | Conditions | Compounds Yield, % | |||
|---|---|---|---|---|---|
| CHTL, M | Time, min | HTL | 2,5-Diketopiperazine | Peptide | |
| Extraction from aqueous solution (pH 10.5) a | 0.40 a | 0.5 a | 91 | 9 | |
| 0.26 | 0.5 | 98 | 1–2 | ||
| 0.20 | 0.5 | 98 | 1–2 | ||
| 0.40 | 10 | 80 | 20 | ||
| 0.26 | 10 | 90 | 10 | ||
| 0.26 | 30 | 60 | 40 b | ||
| Evaporation in vacuo | CHTL = 0.26 M, | ||||
| self-cooling c | 0.5 min | 96 d | 3 | 1 | |
| water bath 25 °C | 0.5 min | 87 | 11 | 2 | |
| water bath 40 °C | 0.5 min | 73 | 24 | 3 | |
| CHTL = 0.26 M, water bath 25 °C | 10 min | 81 | 15 | 5 | |
| CHTL = 0.40 M, water bath 25 °C | 0.5 min | 77 | 15 | 8 | |
| CHTL = 0.40 M, water bath 25 °C | 10 min | 65 | 20 | 15 | |
| Compound storage in solid state | without storage | 96 d | 3 | 1 | |
| −20 °C | 1 day | 94 | 5 | 1 | |
| −20 °C | 1 week | 89 | 9 | 2 | |
| 4 °C | 1 day | 88 | 11 | 1 | |
| 25 °C | 1 day | 75 | 13 | 2 | |
| Conditions | T, °C | Time, h | Compounds Ratio per Perfluoroarene by 19F NMR, % | |||
|---|---|---|---|---|---|---|
 |  |  | Initial PFT or PFX | |||
| PFT 0.01 M, HTL 0.02 M, NEt3 0.04 M | 25 | 20 | 35 | 0 | 0 | 65 |
| 45 | 60 | 5 | 3 | 32 | ||
| 120 | 83 | 11 | 6 | 0 | ||
| PFT 0.02 M, HTL 0.02 M, NEt3 0.04 M | 25 | 20 | 31 | 0 | 0 | 69 |
| 45 | 50 | 2.5 | 0.5 | 47 | ||
| 70 | 61 | 3 | 1 | 35 | ||
| 120 | 83 | 5.5 | 1.5 | 10 | ||
| 150 | 92.5 | 6 | 1.5 | 0 | ||
| PFT 0.01 M, HTL 0.02 M, NEt3 0.08 M | 25 | 20 | 30 | 0 | 0 | 70 |
| 45 | 50 | 3.5 | 1.5 | 45 | ||
| 150 | 87 | 8 | 5 | 0 | ||
| PFT 0.02 M, HTL 0.02 M, NEt3 0.04 M | 50 | 20 | 72 | 2 | 1 | 25 |
| 45 | 88 | 3 | 1.5 | 7.5 | ||
| 70 | 90 | 6.5 | 3.5 | 0 | ||
| PFT 0.009 M, HTL 0.01 M, NEt3 0.013 M | 75 | 20 | 63 | 1 | 0.5 | 35 |
| 45 | 84 | 3 | 1 | 12 | ||
| 70 | 94 | 5 | 1 | 0 | ||
| PFX 0.009 M, HTL 0.01 M, NEt3 0.013 M | 75 | 15 | ~100 | ~0 | 0 | 0 |
| HSA Type | CD Analysis | DLS Analysis | SDS-PAGE Analysis | |||
|---|---|---|---|---|---|---|
| n | α-Helix, % | β-Sheet, % | Hydrodynamic Diameter, nm | Oligomer, % | Monomer, % | |
| HSA | - | 60.0 | 4.5 | 6.1 ± 0.6 1 | 7.0 | 93.0 |
| PFT-HSA | 3.9 ± 0.1 | 56.4 | 6.8 | 6.2 ± 0.8 | 8.7 | 91.3 |
| PFX-HSA | 4.9 ± 0.1 | 58.2 | 5.0 | 6.0 ± 0.9 | 7.5 | 92.5 |
| Hcy-HSA | 2.9 ± 0.1 | 50.0 | 10.0 | n.d. | 83.0 | 17.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mitin, D.E.; Chubarov, A.S. Fluorinated Human Serum Albumin as Potential 19F Magnetic Resonance Imaging Probe. Molecules 2023, 28, 1695. https://doi.org/10.3390/molecules28041695
Mitin DE, Chubarov AS. Fluorinated Human Serum Albumin as Potential 19F Magnetic Resonance Imaging Probe. Molecules. 2023; 28(4):1695. https://doi.org/10.3390/molecules28041695
Chicago/Turabian StyleMitin, Dmitry E., and Alexey S. Chubarov. 2023. "Fluorinated Human Serum Albumin as Potential 19F Magnetic Resonance Imaging Probe" Molecules 28, no. 4: 1695. https://doi.org/10.3390/molecules28041695
APA StyleMitin, D. E., & Chubarov, A. S. (2023). Fluorinated Human Serum Albumin as Potential 19F Magnetic Resonance Imaging Probe. Molecules, 28(4), 1695. https://doi.org/10.3390/molecules28041695