

Virtual Screening of Hepatitis B Virus Pre-Genomic RNA as a Novel Therapeutic Target
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
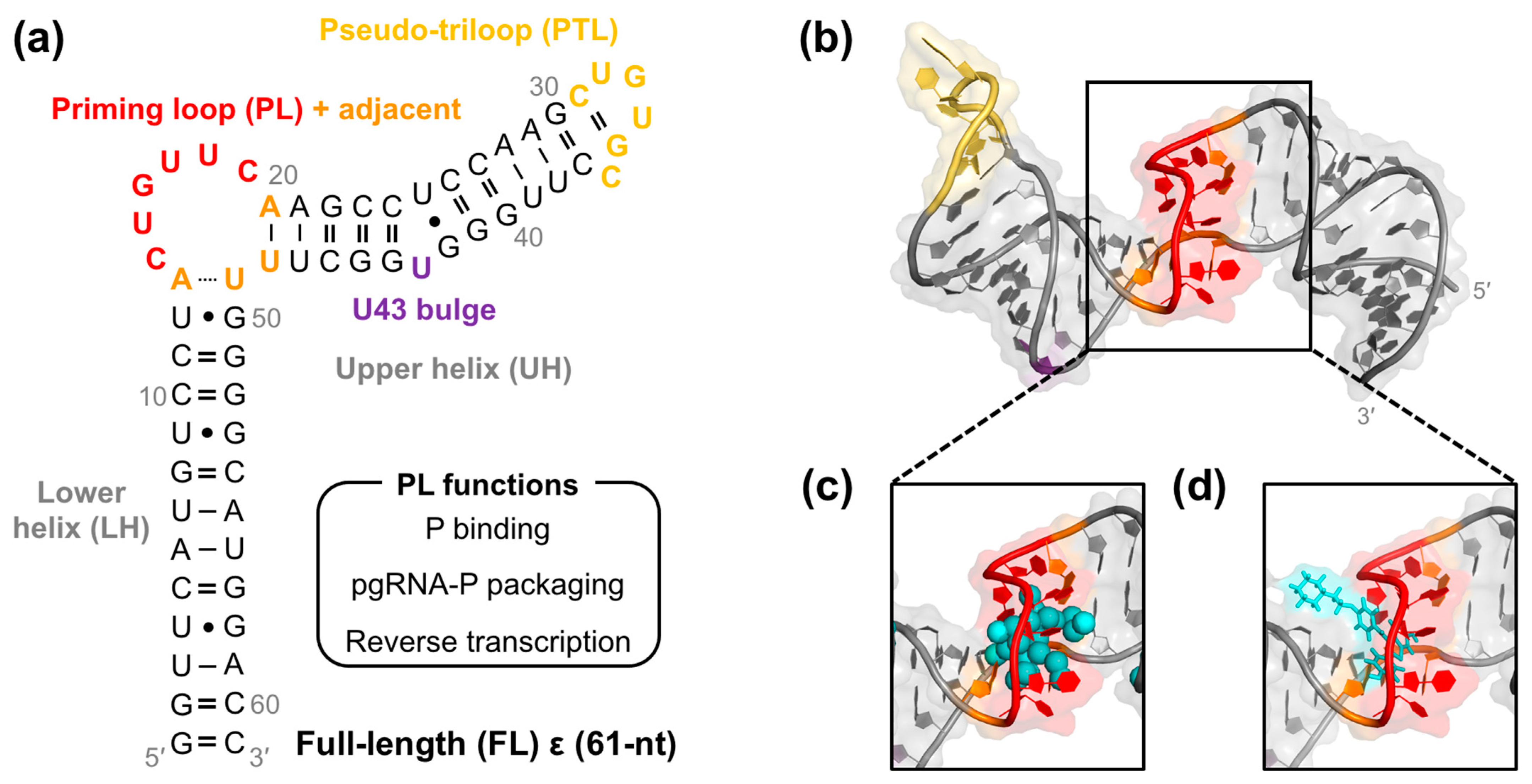
2. Results
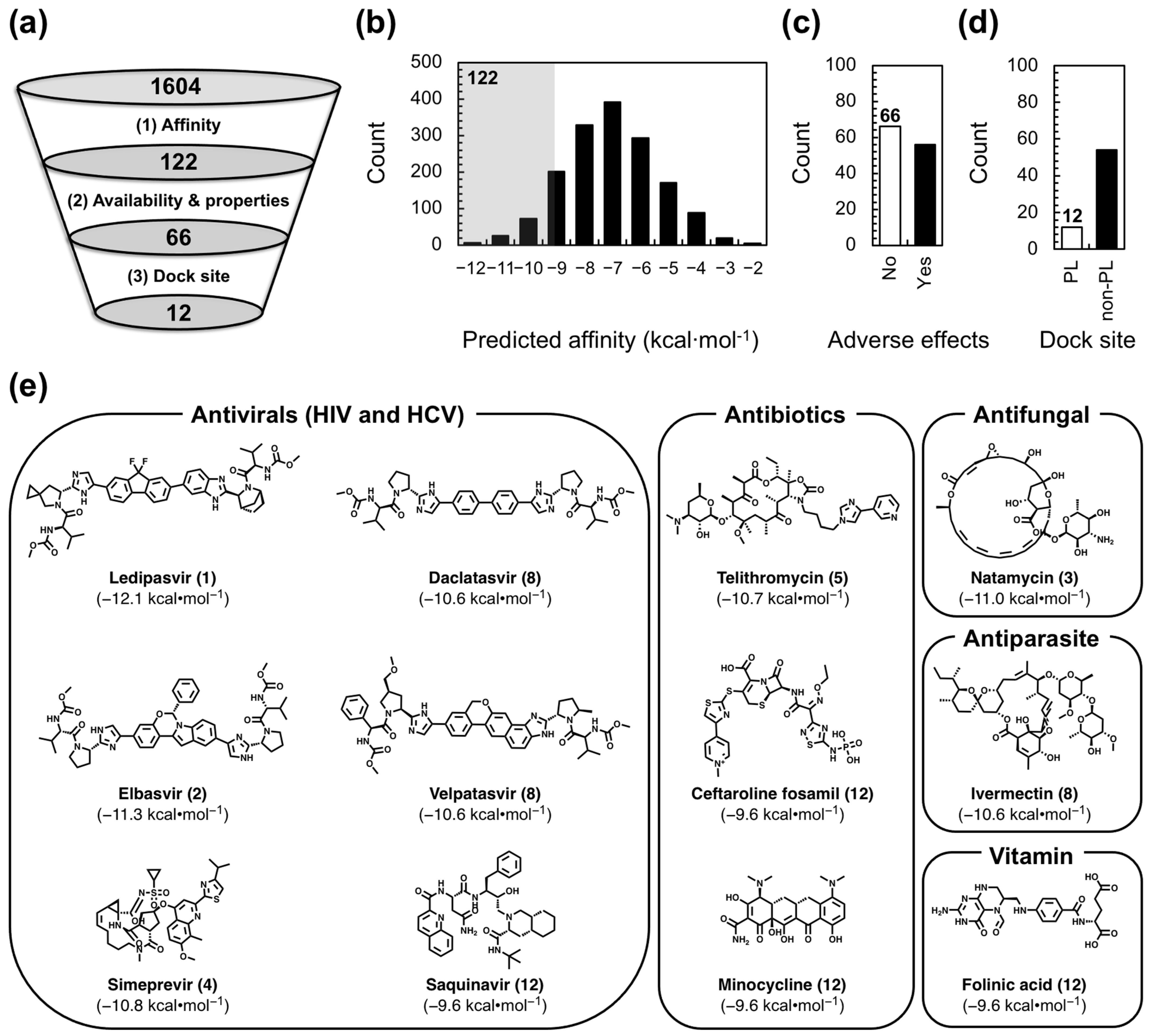
2.1. Virtual Screen Strategy
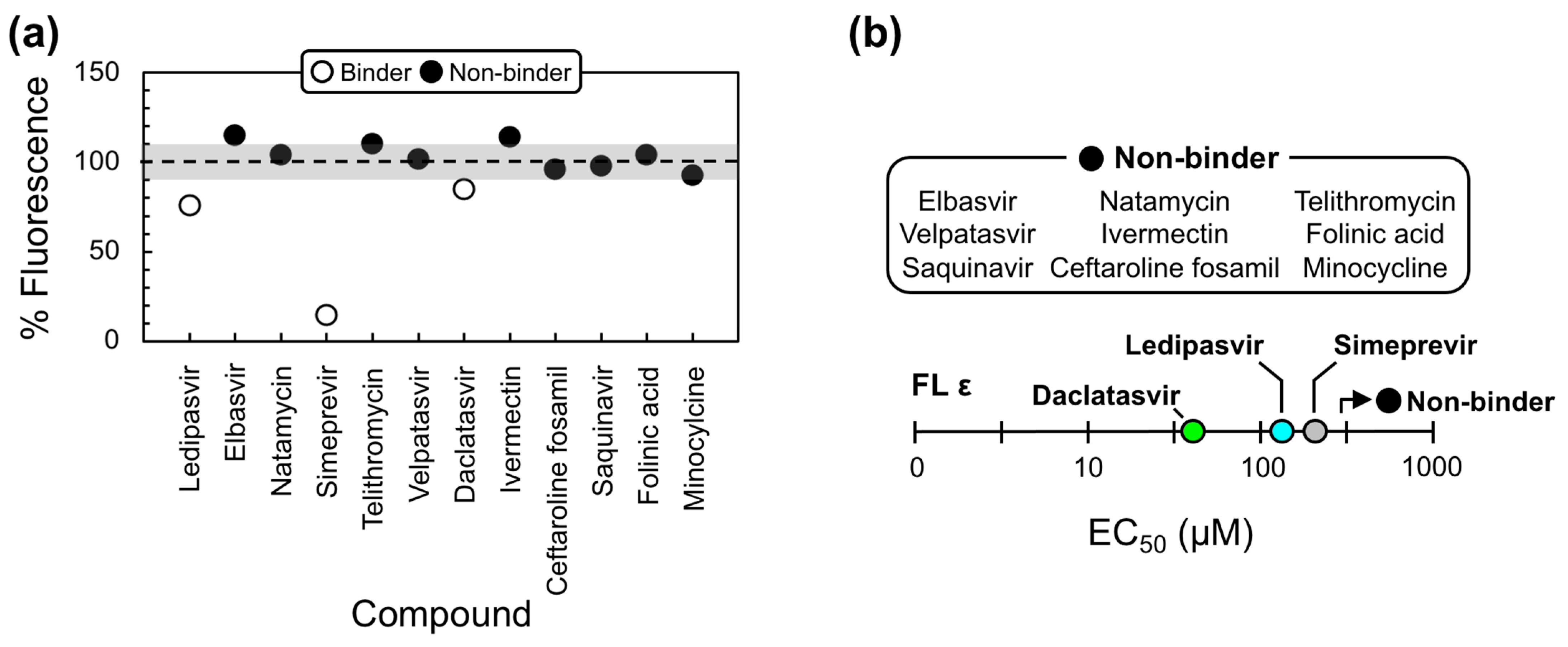
2.2. Lead Compound Generation
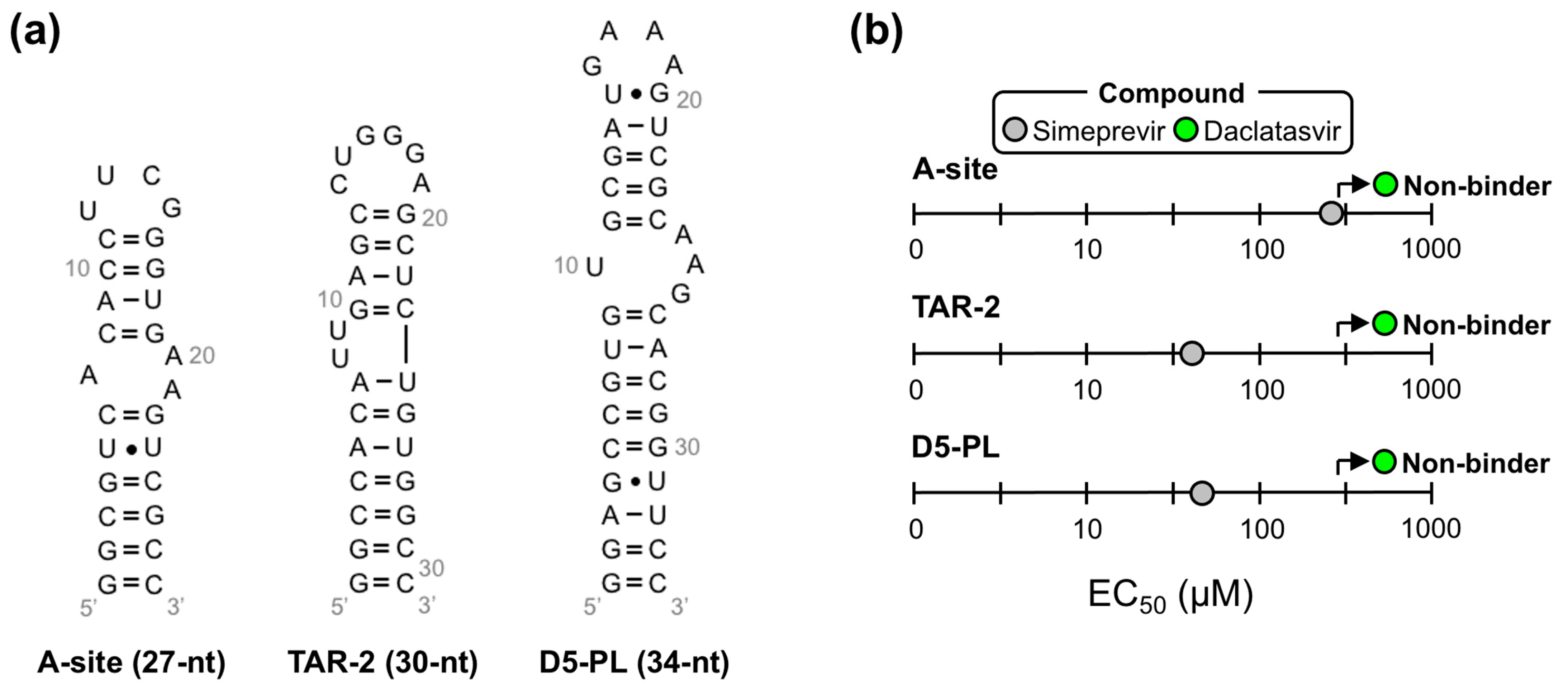
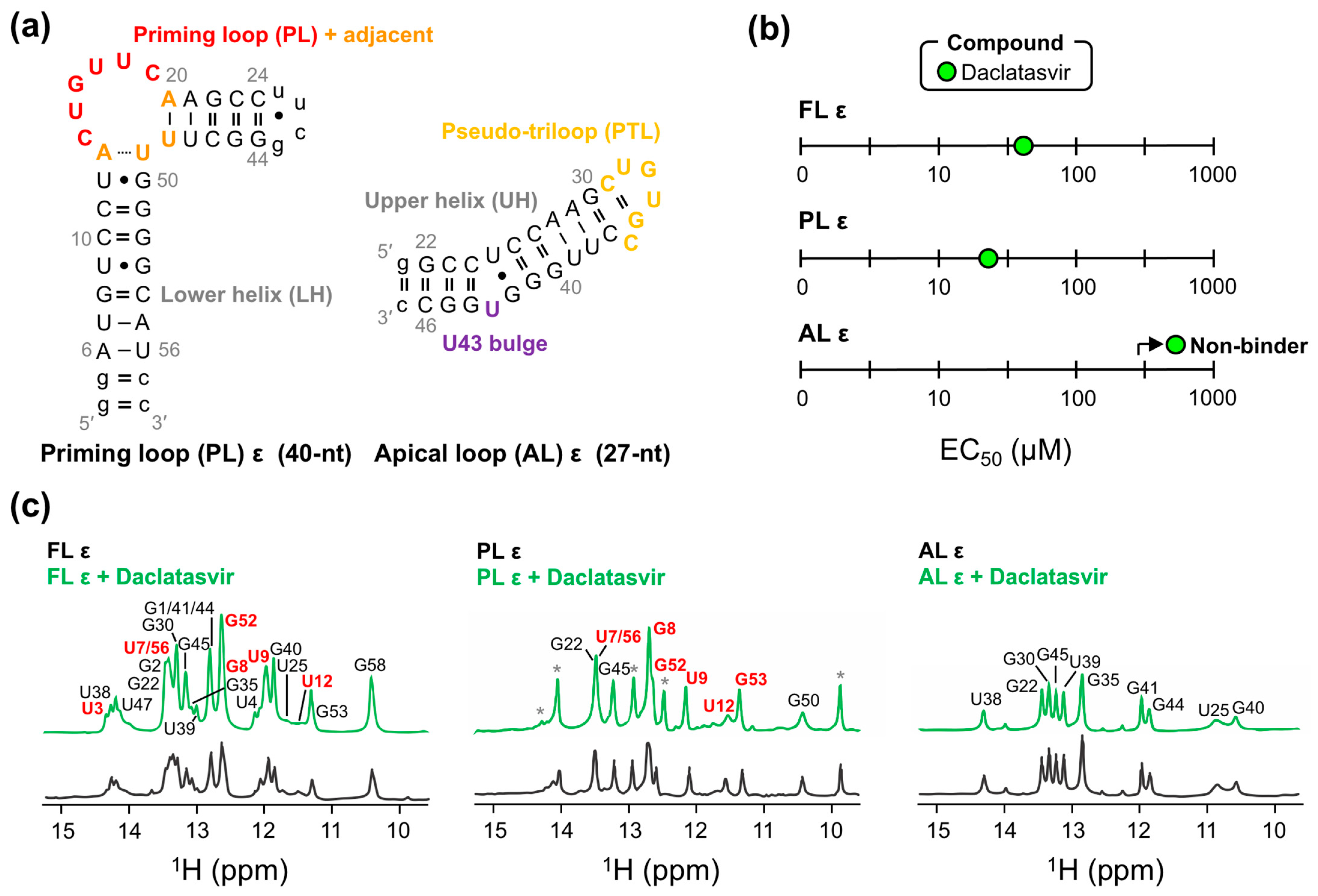
2.3. Daclatasvir Selectively Targets the ε Priming Loop
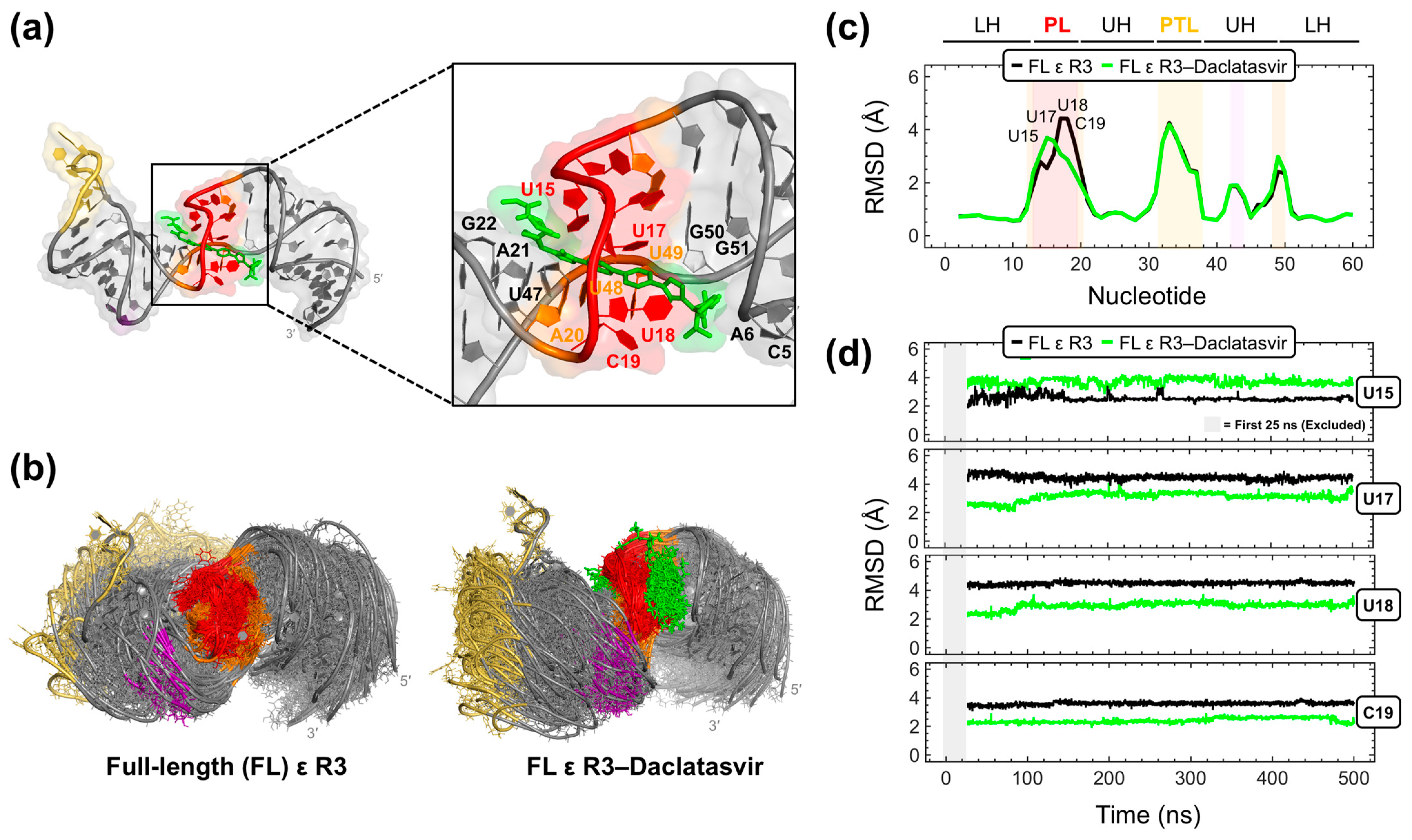
2.4. Modeling the Full-Length ε-Daclatasvir Complex
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Virtual Screening
4.3. RNA Transcription
4.4. Dye-Displacement Assay
4.5. NMR Titrations
4.6. Computational Docking
4.7. Molecular Dynamic Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Sheena, B.S.; Hiebert, L.; Han, H.; Ippolito, H.; Abbasi-Kangevari, M.; Abbasi-Kangevari, Z.; Abbastabar, H.; Abdoli, A.; Abubaker Ali, H.; Adane, M.M.; et al. Global, regional, and national burden of hepatitis B, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet Gastroenterol. Hepatol. 2022, 7, 796–829. [Google Scholar] [CrossRef]
- Perz, J.F.; Armstrong, G.L.; Farrington, L.A.; Hutin, Y.J.F.; Bell, B.P. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J. Hepatol. 2006, 45, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Woo, A.S.J.; Kwok, R.; Ahmed, T. Alpha-interferon treatment in hepatitis B. Ann. Transl. Med. 2017, 5, 159. [Google Scholar] [CrossRef]
- de Clercq, E.; Férir, G.; Kaptein, S.; Neyts, J. Antiviral Treatment of Chronic Hepatitis B Virus (HBV) Infections. Viruses 2010, 2, 1279–1305. [Google Scholar] [CrossRef]
- Zoulim, F.; Locarnini, S. Hepatitis B virus resistance to nucleos(t)ide analogues. Gastroenterology 2009, 137, 1593–1608.e1–2. [Google Scholar] [CrossRef]
- Menéndez-Arias, L.; Álvarez, M.; Pacheco, B. Nucleoside/nucleotide analog inhibitors of hepatitis B virus polymerase: Mechanism of action and resistance. Curr. Opin. Virol. 2014, 8, 1–9. [Google Scholar] [CrossRef]
- Scaglione, S.J.; Lok, A.S.F. Effectiveness of hepatitis B treatment in clinical practice. Gastroenterology 2012, 142, 1360–1368.e1. [Google Scholar] [CrossRef]
- Robinson, W.S.; Clayton, D.A.; Greenman, R.L. DNA of a human hepatitis B virus candidate. J. Virol. 1974, 14, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Pasek, M.; Goto, T.; Gilbert, W.; Zink, B.; Schaller, H.; Mackay, P.; Leadbetter, G.; Murray, K. Hepatitis B virus genes and their expression in E. coli. Nature 1979, 282, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Charnay, P.; Mandart, E.; Hampe, A.; Fitoussi, F.; Tiollais, P.; Galibert, F. Localization on the viral genome and nucleotide sequence of the gene coding for the two major polypeptides of the hepatitis B surface antigen (HBs Ag). Nucleic Acids Res. 1979, 7, 335–346. [Google Scholar] [CrossRef] [Green Version]
- Valenzuela, P.; Gray, P.; Quiroga, M.; Zaldivar, J.; Goodman, H.M.; Rutter, W.J. Nucleotide sequence of the gene coding for the major protein of hepatitis B virus surface antigen. Nature 1979, 280, 815–819. [Google Scholar] [CrossRef]
- Galibert, F.; Mandart, E.; Fitoussi, F.; Tiollais, P.; Charnay, P. Nucleotide sequence of the hepatitis B virus genome (subtype ayw) cloned in E. coli. Nature 1979, 281, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Junker-Niepmann, M.; Schaller, H. The P gene product of hepatitis B virus is required as a structural component for genomic RNA encapsidation. J. Virol. 1990, 64, 5324–5332. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.A.; Hu, J. Hepatitis B virus reverse transcriptase: Diverse functions as classical and emerging targets for antiviral intervention. Emerg. Microbes Infect. 2013, 2, e56. [Google Scholar] [CrossRef] [PubMed]
- Radziwill, G.; Tucker, W.; Schaller, H. Mutational analysis of the hepatitis B virus P gene product: Domain structure and RNase H activity. J. Virol. 1990, 64, 613–620. [Google Scholar] [CrossRef] [PubMed]
- Toh, H.; Hayashida, H.; Miyata, T. Sequence homology between retroviral reverse transcriptase and putative polymerases of hepatitis B virus and cauliflower mosaic virus. Nature 1983, 305, 827–829. [Google Scholar] [CrossRef] [PubMed]
- Zoulim, F.; Seeger, C. Reverse transcription in hepatitis B viruses is primed by a tyrosine residue of the polymerase. J. Virol. 1994, 68, 6–13. [Google Scholar] [CrossRef]
- Weber, W.; Bronsema, V.; Bartos, H.; Bosserhoff, A.; Bartenschlager, R.; Schaller, H. Hepadnavirus P protein utilizes a tyrosine residue in the TP domain to prime reverse transcription. J. Virol. 1994, 68, 2994–2999. [Google Scholar] [CrossRef]
- Wang, G.H.; Seeger, C. The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. Cell 1992, 71, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Junker-Niepmann, M.; Bartenschlager, R.; Schaller, H. A short cis-acting sequence is required for hepatitis B virus pregenome encapsidation and sufficient for packaging of foreign RNA. EMBO J. 1990, 9, 3389–3396. [Google Scholar] [CrossRef]
- Wang, G.-H.; Zoulim, F.; Leber, E.H.; Kitson, J.; Seeger, C. Role of RNA in enzymatic activity of the reverse transcriptase of hepatitis B viruses. J. Virol. 1994, 68, 8437–8442. [Google Scholar] [CrossRef]
- Knaus, T.; Nassal, M. The encapsidation signal on the hepatitis B virus RNA pregenome forms a stem-loop structure that is critical for its function. Nucleic Acids Res. 1993, 21, 3967–3975. [Google Scholar] [CrossRef]
- Pollack, J.R.; Ganem, D. An RNA stem-loop structure directs hepatitis B virus genomic RNA encapsidation. J. Virol. 1993, 67, 3254–3263. [Google Scholar] [CrossRef]
- Hirsch, R.C.; Lavine, J.E.; Chang, L.J.; Varmus, H.E.; Ganem, D. Polymerase gene products of hepatitis B viruses are required for genomic RNA packaging as wel as for reverse transcription. Nature 1990, 344, 552–555. [Google Scholar] [CrossRef]
- Bartenschlager, R.; Schaller, H. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J. 1992, 11, 3413–3420. [Google Scholar] [CrossRef]
- Reiger, A.; Nassal, M. Specific hepatitis B virus minus-strand DNA synthesis requires only the 5′ encapsidation signal and the 3′-proximal direct repeat DR1. J. Virol. 1996, 70, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Fallows, D.A.; Goff, S.P. Mutations in the epsilon sequences of human hepatitis B virus affect both RNA encapsidation and reverse transcription. J. Virol. 1995, 69, 3067–3073. [Google Scholar] [CrossRef] [PubMed]
- Lanford, R.E.; Notvall, L.; Beames, B. Nucleotide priming and reverse transcriptase activity of hepatitis B virus polymerase expressed in insect cells. J. Virol. 1995, 69, 4431–4439. [Google Scholar] [CrossRef] [PubMed]
- Nassal, M.; Rieger, A. A bulged region of the hepatitis B virus RNA encapsidation signal contains the replication origin for discontinuous first-strand DNA synthesis. J. Virol. 1996, 70, 2764–2773. [Google Scholar] [CrossRef]
- LeBlanc, R.M.; Kasprzak, W.K.; Longhini, A.P.; Olenginski, L.T.; Abulwerdi, F.; Ginocchio, S.; Shields, B.; Nyman, J.; Svirydava, M.; Del Vecchio, C.; et al. Structural insights of the conserved “priming loop” of hepatitis B virus pre-genomic RNA. J. Biomol. Struct. Dyn. 2022, 40, 9761–9773. [Google Scholar] [CrossRef]
- Xie, J.; Frank, A.T. Mining for Ligandable Cavities in RNA. ACS Med. Chem. Lett. 2021, 12, 928–934. [Google Scholar] [CrossRef]
- Olenginski, L.T.; Kasprzak, W.K.; Bergonzo, C.; Shapiro, B.A.; Dayie, T.K. Conformational dynamics of the hepatitis B virus pre-genomic RNA on multiple time scales: Implications for viral replication. J. Mol. Biol. 2022, 434, 167633. [Google Scholar] [CrossRef]
- Jones, S.A.; Boregowda, R.; Spratt, T.E.; Hu, J. In vitro epsilon RNA-dependent protein priming activity of human hepatitis B virus polymerase. J. Virol. 2012, 86, 5134–5150. [Google Scholar] [CrossRef]
- Hu, J.; Boyer, M. Hepatitis B virus reverse transcriptase and ε RNA sequences required for specific interaction in vitro. J. Virol. 2006, 80, 2141–2150. [Google Scholar] [CrossRef]
- Bak, E.; Miller, J.T.; Noronha, A.; Tavis, J.; Gallicchio, E.; Murelli, R.P.; Le Grice, S.F.J. 3,7-Dihydroxytropolones Inhibit Initiation of Hepatitis B Virus Minus-Strand DNA Synthesis. Molecules 2020, 25, 4434. [Google Scholar] [CrossRef]
- Knegtel, R.M.A.; Kuntz, I.D.; Oshiro, C.M. Molecular docking to ensembles of protein structures. J. Mol. Biol. 1997, 266, 424–440. [Google Scholar] [CrossRef]
- Lin, J.H.; Perryman, A.L.; Schames, J.R.; McCammon, J.A. Computational drug design accommodating receptor flexibility: The relaxed complex scheme. J. Am. Chem. Soc. 2002, 124, 5632–5633. [Google Scholar] [CrossRef]
- Carlson, H.A.; Masukawa, K.M.; Rubins, K.; Bushman, F.D.; Jorgensen, W.L.; Lins, R.D.; Briggs, J.M.; McCammon, J.A. Developing a dynamic pharmacophore model for HIV-1 integrase. J. Med. Chem. 2000, 43, 2100–2114. [Google Scholar] [CrossRef]
- Stelzer, A.C.; Frank, A.T.; Kratz, J.D.; Swanson, M.D.; Gonzalez-Hernandez, M.J.; Lee, J.; Andricioaei, I.; Markovitz, D.M.; Al-Hashimi, H.M. Discovery of selective bioactive small molecules by targeting an RNA dynamic ensemble. Nat. Chem. Biol. 2011, 7, 553–559. [Google Scholar] [CrossRef]
- Ganser, L.R.; Lee, J.; Rangadurai, A.; Merriman, D.K.; Kelly, M.L.; Kansal, A.D.; Sathyamoorthy, B.; Al-Hashimi, H.M. High-performance virtual screening by targeting a high-resolution RNA dynamic ensemble. Nat. Struct. Mol. Biol. 2018, 25, 425–434. [Google Scholar] [CrossRef] [Green Version]
- Frank, A.T.; Stelzer, A.C.; Al-Hashimi, H.M.; Andricioaei, I. Constructing RNA dynamical ensembles by combining MD and motionally decoupled NMR RDCs: New insights into RNA dynamics and adaptive ligand recognition. Nucleic Acids Res. 2009, 37, 3670–3679. [Google Scholar] [CrossRef] [PubMed]
- Salmon, L.; Bascom, G.; Andricioaei, I.; Al-Hashimi, H.M. A general method for constructing atomic-resolution RNA ensembles using NMR residual dipolar couplings: The basis for interhelical motions revealed. J. Am. Chem. Soc. 2013, 135, 5457–5466. [Google Scholar] [CrossRef]
- Salmon, L.; Yang, S.; Al-Hashimi, H.M. Advances in the determination of nucleic acid conformational ensembles. Annu. Rev. Phys. Chem. 2014, 65, 293–316. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Coleman, R.G.; Fraser, J.S.; Shoichet, B.K. Incorporation of protein flexibility and conformational energy penalties in docking screens to improve ligand discovery. Nat. Chem. 2014, 6, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Tóth, G.; Gardai, S.J.; Zago, W.; Bertoncini, C.W.; Cremades, N.; Roy, S.L.; Tambe, M.A.; Rochet, J.C.; Galvagnion, C.; Skibinski, G.; et al. Targeting the intrinsically disordered structural ensemble of α-synuclein by small molecules as a potential therapeutic strategy for Parkinson’s disease. PLoS ONE 2014, 9, e87133. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15—Ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC—A free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar]
- Balani, S.; Miwa, G.; Gan, L.-S.; Wu, J.-T.; Lee, F. Strategy of utilizing in vitro and in vivo ADME tools for lead optimization and drug candidate selection. Curr. Top. Med. Chem. 2005, 5, 1033–1038. [Google Scholar] [CrossRef]
- Singh, S. Preclinical pharmacokinetics: An approach towards safer and efficacious drugs. Curr. Drug Metab. 2006, 7, 165–182. [Google Scholar] [CrossRef]
- Tetko, I.V.; Bruneau, P.; Mewes, H.W.; Rohrer, D.C.; Poda, G.I. Can we estimate the accuracy of ADME-Tox predictions? Drug Discov. Today 2006, 11, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today. Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Dallakyan, S.; Olson, A.J. Small-molecule library screening by docking with PyRx. Methods Mol. Biol. 2015, 1263, 243–250. [Google Scholar]
- Zhou, Y.; Jiang, Y.; Chen, S.J. RNA-ligand molecular docking: Advances and challenges. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2022, 12, e1571. [Google Scholar] [CrossRef]
- Ruiz-Carmona, S.; Alvarez-Garcia, D.; Foloppe, N.; Garmendia-Doval, A.B.; Juhos, S.; Schmidtke, P.; Barril, X.; Hubbard, R.E.; Morley, S.D. rDock: A fast, versatile and open source program for docking ligands to proteins and nucleic acids. PLoS Comput. Biol. 2014, 10, e1003571. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, S.; Xie, J.; Frank, A.T. RNAPosers: Machine learning classifiers for ribonucleic acid-ligand poses. J. Phys. Chem. B 2020, 124, 4436–4445. [Google Scholar] [CrossRef] [PubMed]
- Kammarabutr, J.; Mahalapbutr, P.; Okumura, H.; Wolschann, P.; Rungrotmongkol, T. Structural dynamics and susceptibility of anti-HIV drugs against HBV reverse transcriptase. J. Biomol. Struct. Dyn. 2021, 39, 2502–2511. [Google Scholar] [CrossRef]
- Das, K.; Xiong, X.; Yang, H.; Westland, C.E.; Gibbs, C.S.; Sarafianos, S.G.; Arnold, E. Molecular modeling and biochemical characterization reveal the mechanism of hepatitis B virus polymerase resistance to lamivudine (3TC) and emtricitabine (FTC). J. Virol. 2001, 75, 4771–4779. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Thai, H.; Kitrinos, K.M.; Xia, G.; Gaggar, A.; Paulson, M.; Ganova-Raeva, L.; Khudyakov, Y.; Lara, J. Modeling the functional state of the reverse transcriptase of hepatitis B virus and its application to probing drug-protein interaction. BMC Bioinform. 2016, 17, 280. [Google Scholar] [CrossRef] [PubMed]
- Buhlig, T.S.; Bowersox, A.F.; Braun, D.L.; Owsley, D.N.; James, K.D.; Aranda, A.J.; Kendrick, C.D.; Skalka, N.A.; Clark, D.N. Molecular, evolutionary, and structural analysis of the terminal protein domain of hepatitis B virus polymerase, a potential drug rarget. Viruses 2020, 12, 570. [Google Scholar] [CrossRef] [PubMed]
- Tajwar, R.; Bradley, D.P.; Ponzar, N.L.; Tavis, J.E. Predicted structure of the hepatitis B virus polymerase reveals an ancient conserved protein fold. Protein Sci. 2022, 31, e4421. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Milligan, J.F.; Uhlenbeck, O.C. Synthesis of small RNAs using T7 RNA polymerase. Methods Enzymol. 1989, 180, 51–62. [Google Scholar] [PubMed]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Bergonzo, C.; Cheatham, T.E. Improved force field parameters lead to a better description of RNA structure. J. Chem. Theory Comput. 2015, 11, 3969–3972. [Google Scholar] [CrossRef]
- Zgarbová, M.; Otyepka, M.; Šponer, J.; Mládek, A.; Banáš, P.; Cheatham, T.E.; Jurečka, P. Refinement of the Cornell et al. Nucleic acids force field based on reference quantum chemical calculations of glycosidic torsion profiles. J. Chem. Theory Comput. 2011, 7, 2886–2902. [Google Scholar] [CrossRef]
- Steinbrecher, T.; Latzer, J.; Case, D.A. Revised AMBER parameters for bioorganic phosphates. J. Chem. Theory Comput. 2012, 8, 4405–4412. [Google Scholar] [CrossRef]
- Mukhopadhyay, A.; Fenley, A.T.; Tolokh, I.S.; Onufriev, A.V. Charge hydration asymmetry: The basic principle and how to use it to test and improve water models. J. Phys. Chem. B 2012, 116, 9776–9783. [Google Scholar] [CrossRef]
- Izadi, S.; Anandakrishnan, R.; Onufriev, A.V. Building water models: A different approach. J. Phys. Chem. Lett. 2014, 5, 3863–3871. [Google Scholar] [CrossRef] [PubMed]
- Joung, I.S.; Cheatham, T.E. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheatham, T.E.; Miller, J.L.; Fox, T.; Darden, T.A.; Kollman, P.A. Molecular Dynamics Simulations on Solvated Biomolecular Systems: The particle mesh Ewald method leads to stable trajectories of DNA, RNA, and proteins. J. Am. Chem. Soc. 1995, 117, 4193–4194. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1998, 81, 3684. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olenginski, L.T.; Kasprzak, W.K.; Attionu, S.K.; Shapiro, B.A.; Dayie, T.K. Virtual Screening of Hepatitis B Virus Pre-Genomic RNA as a Novel Therapeutic Target. Molecules 2023, 28, 1803. https://doi.org/10.3390/molecules28041803
Olenginski LT, Kasprzak WK, Attionu SK, Shapiro BA, Dayie TK. Virtual Screening of Hepatitis B Virus Pre-Genomic RNA as a Novel Therapeutic Target. Molecules. 2023; 28(4):1803. https://doi.org/10.3390/molecules28041803
Chicago/Turabian StyleOlenginski, Lukasz T., Wojciech K. Kasprzak, Solomon K. Attionu, Bruce A. Shapiro, and Theodore K. Dayie. 2023. "Virtual Screening of Hepatitis B Virus Pre-Genomic RNA as a Novel Therapeutic Target" Molecules 28, no. 4: 1803. https://doi.org/10.3390/molecules28041803
APA StyleOlenginski, L. T., Kasprzak, W. K., Attionu, S. K., Shapiro, B. A., & Dayie, T. K. (2023). Virtual Screening of Hepatitis B Virus Pre-Genomic RNA as a Novel Therapeutic Target. Molecules, 28(4), 1803. https://doi.org/10.3390/molecules28041803