

Understanding of Active Sites and Interconversion of Pd and PdO during CH4 Oxidation


 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results and Discussion
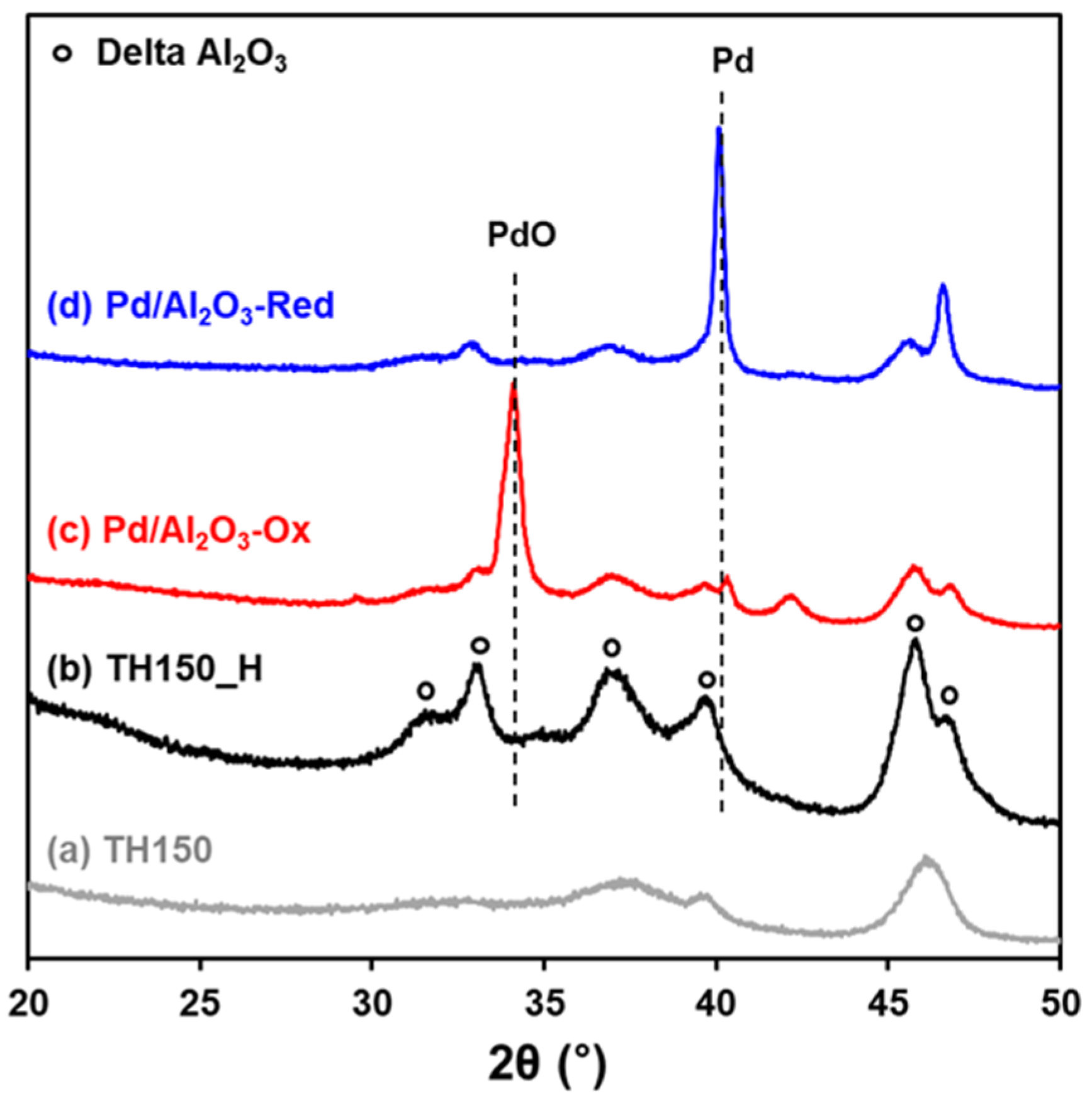
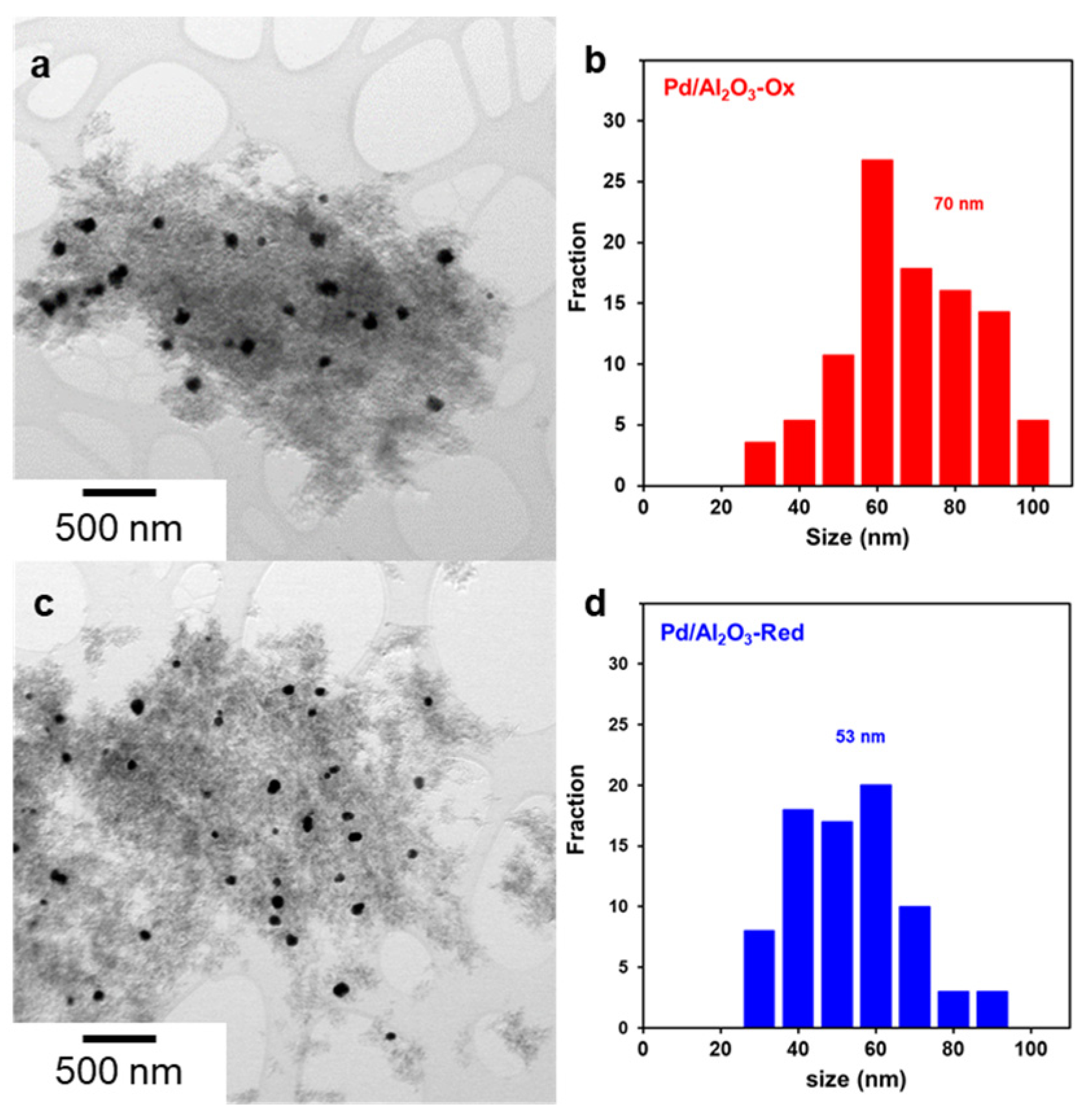
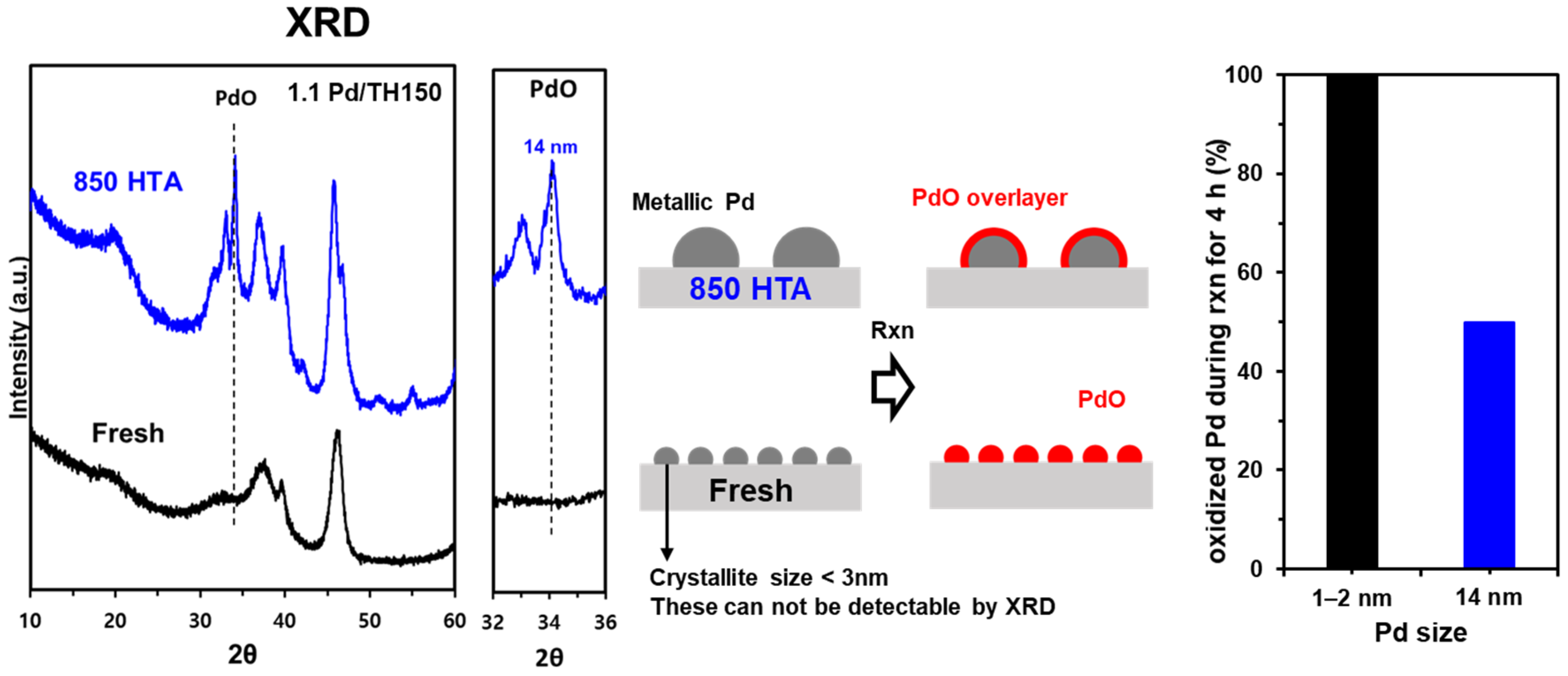
2.1. Characterization of Pd/Al2O3 Catalysts
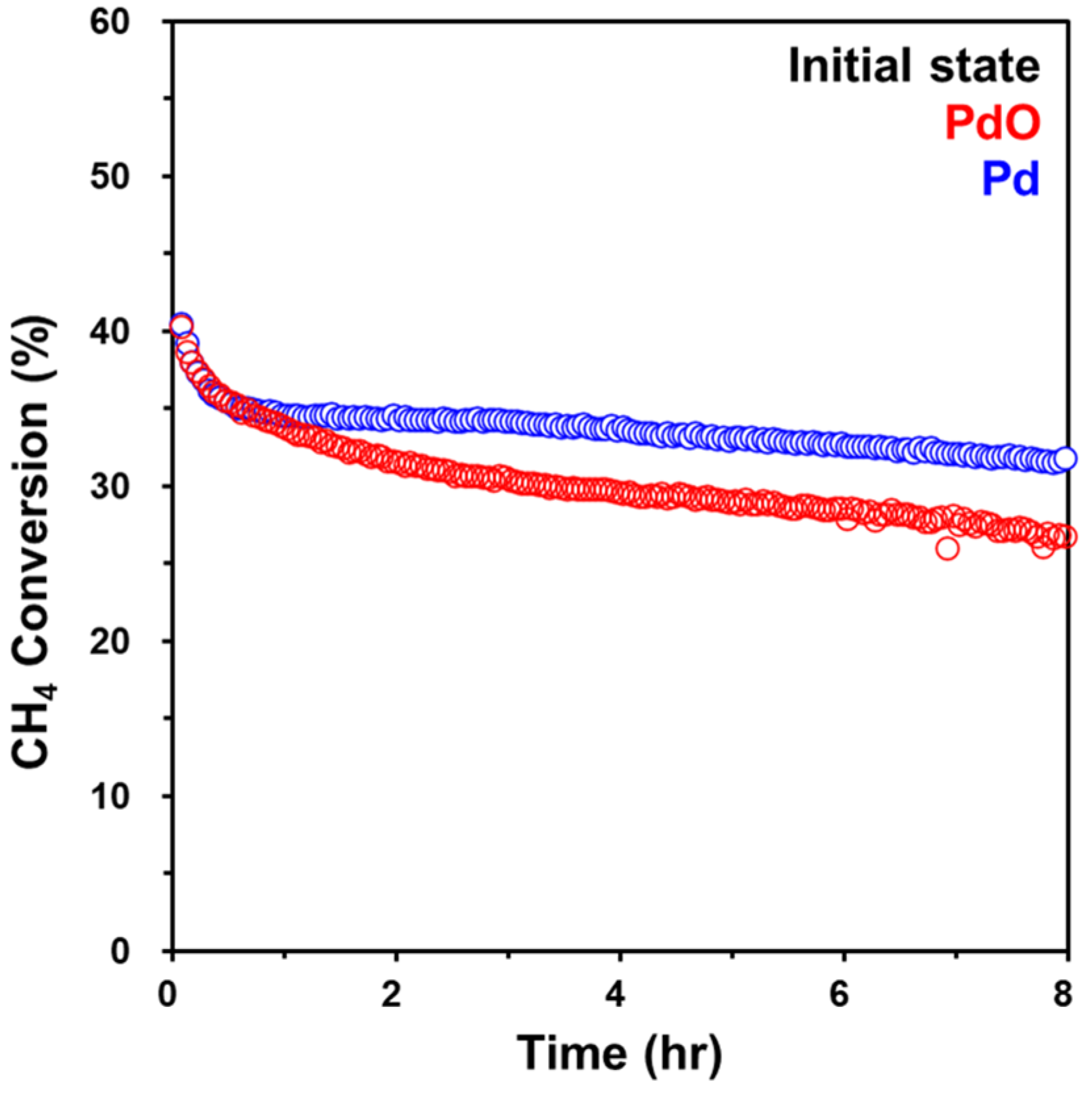
2.2. Catalytic Behaviors of Pd/Al2O3 Catalysts
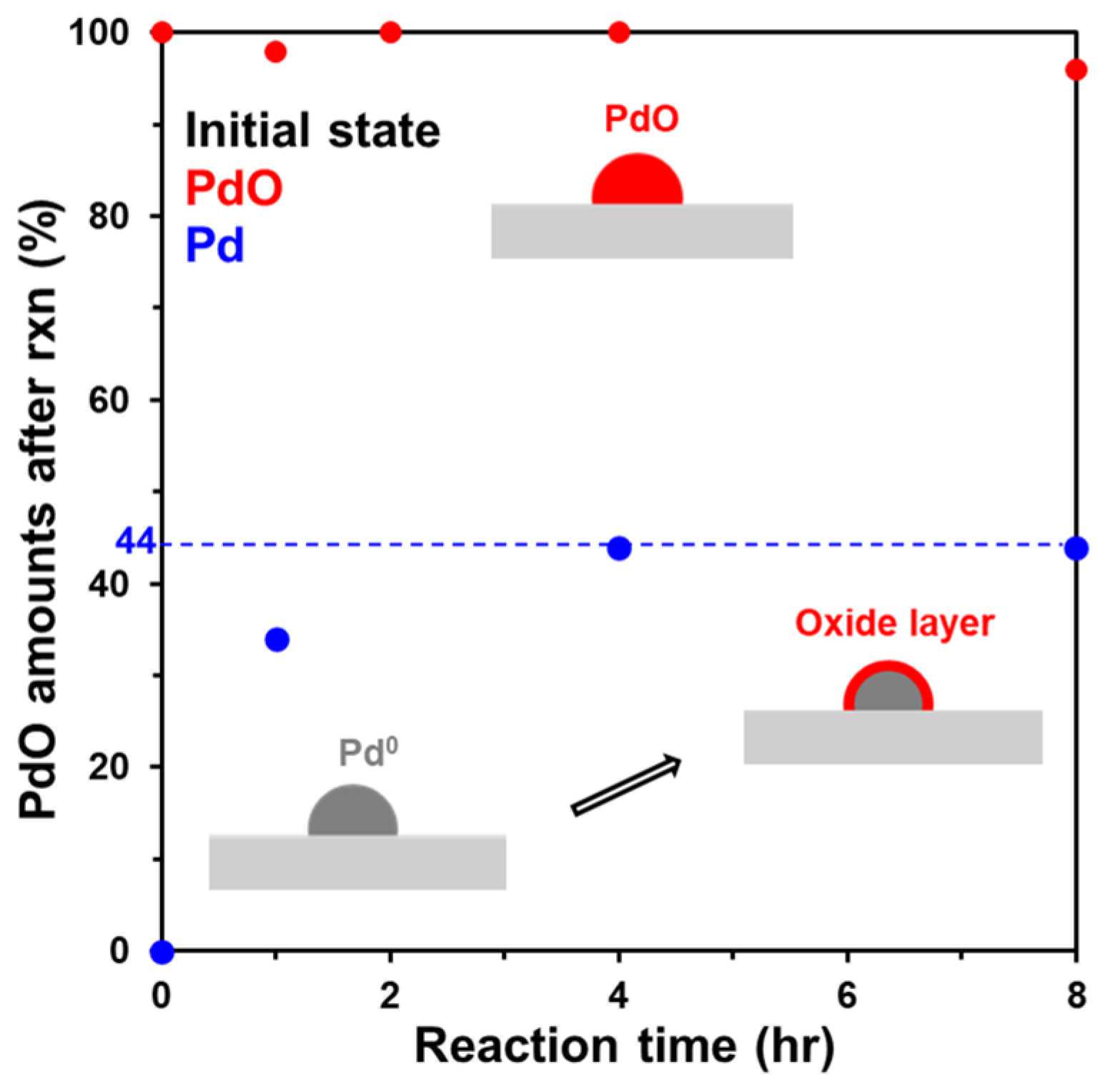
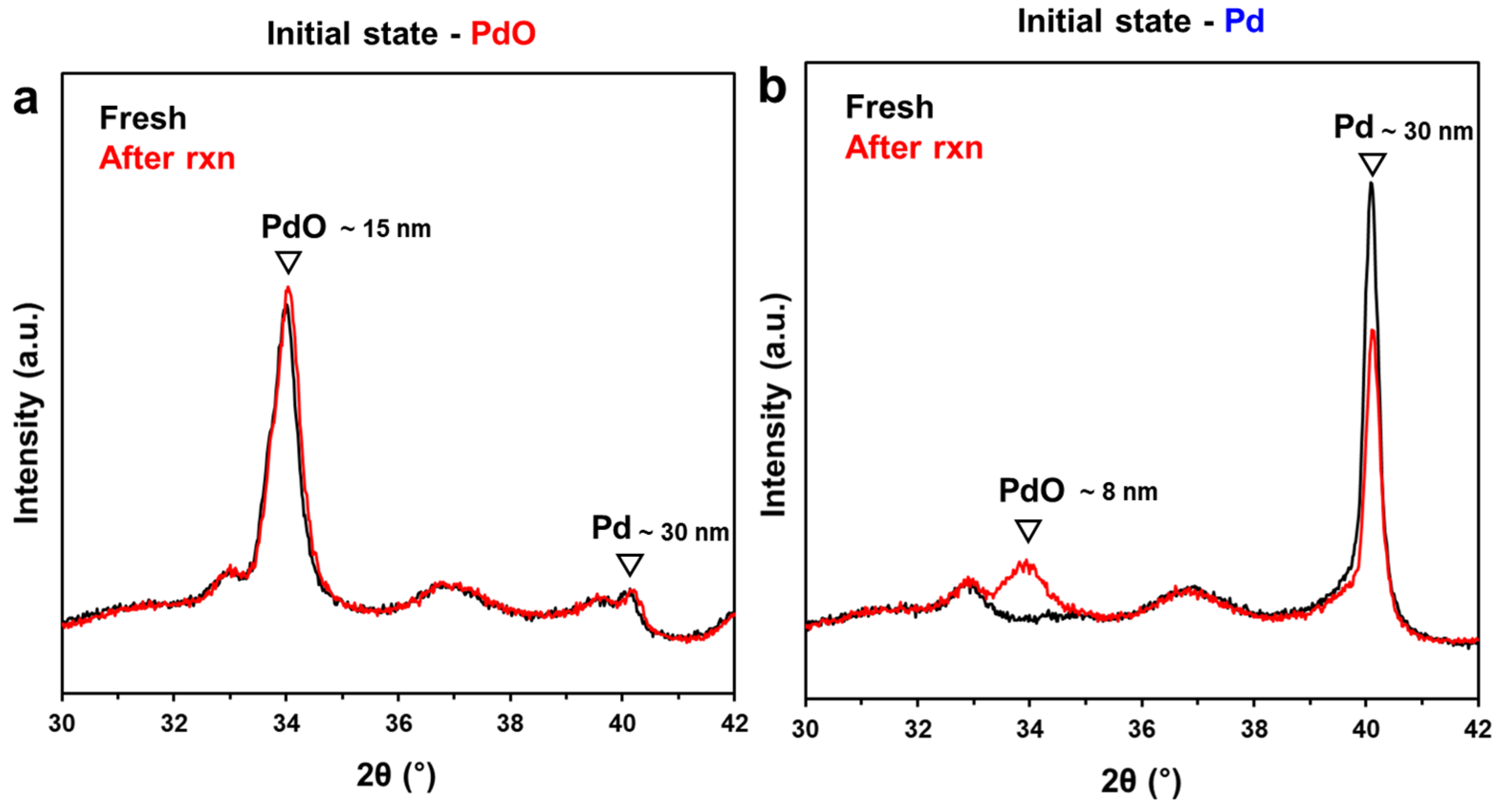
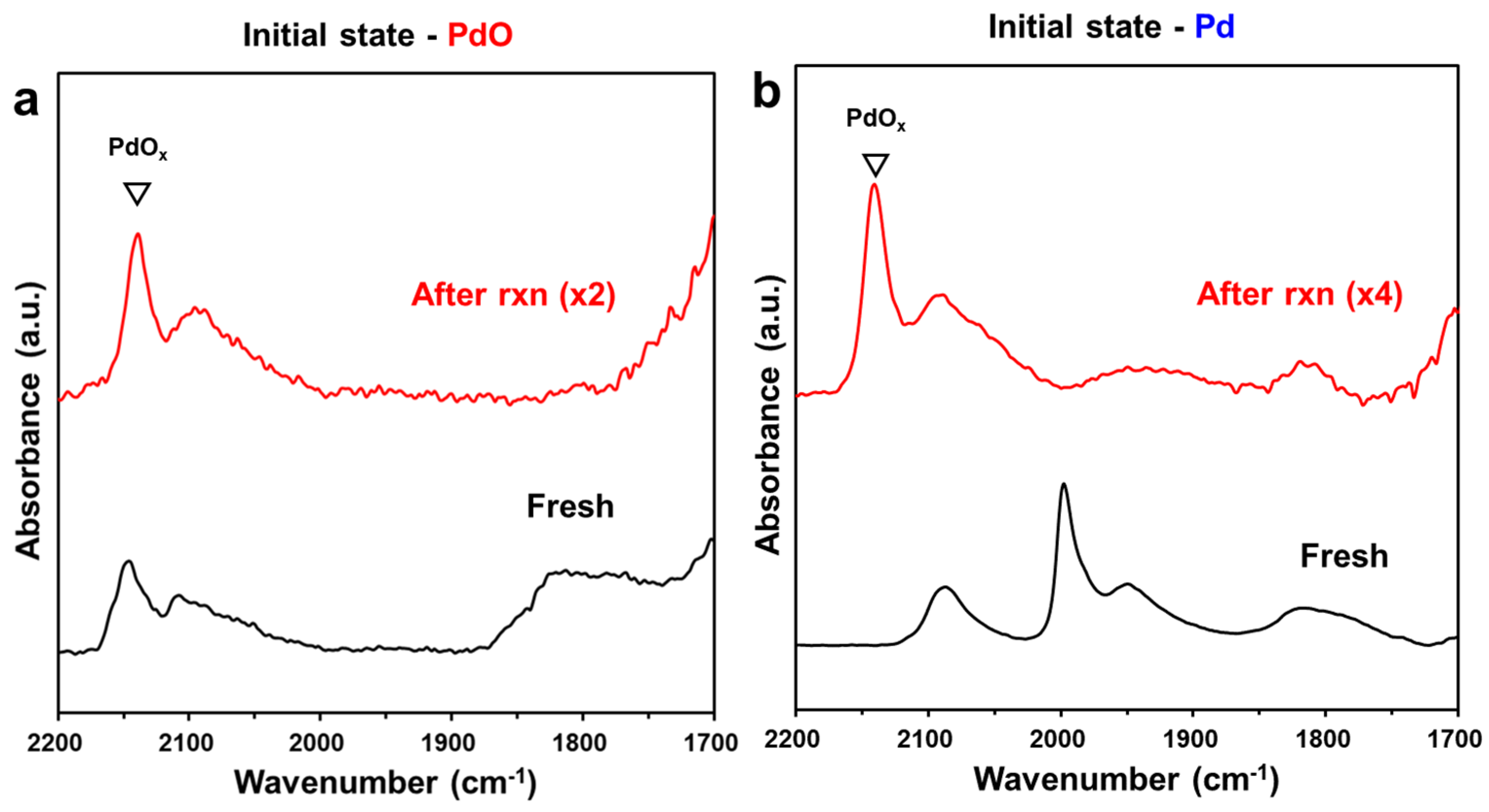
2.3. Comparison of Pd and PdO under the Reaction Conditions
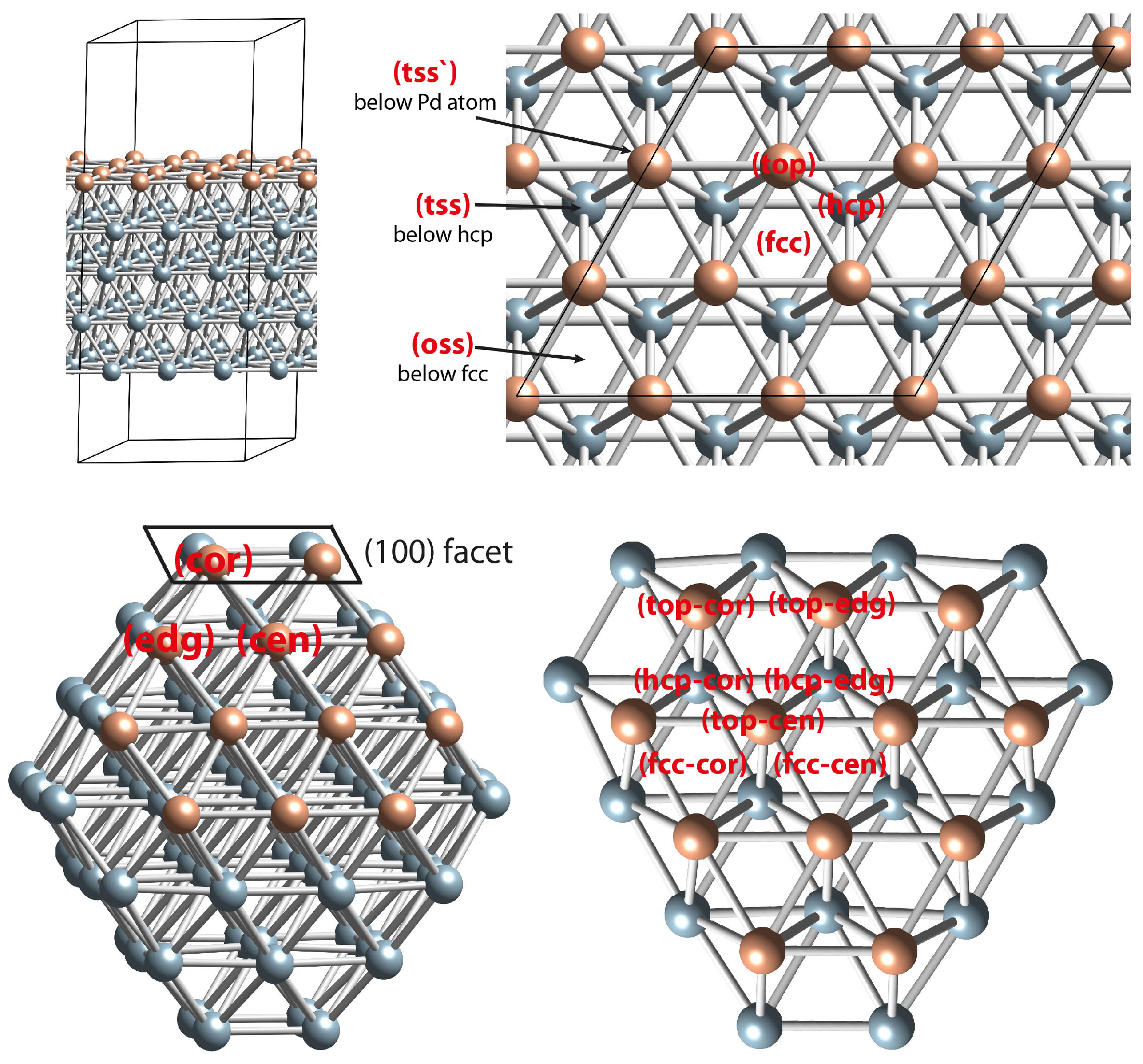
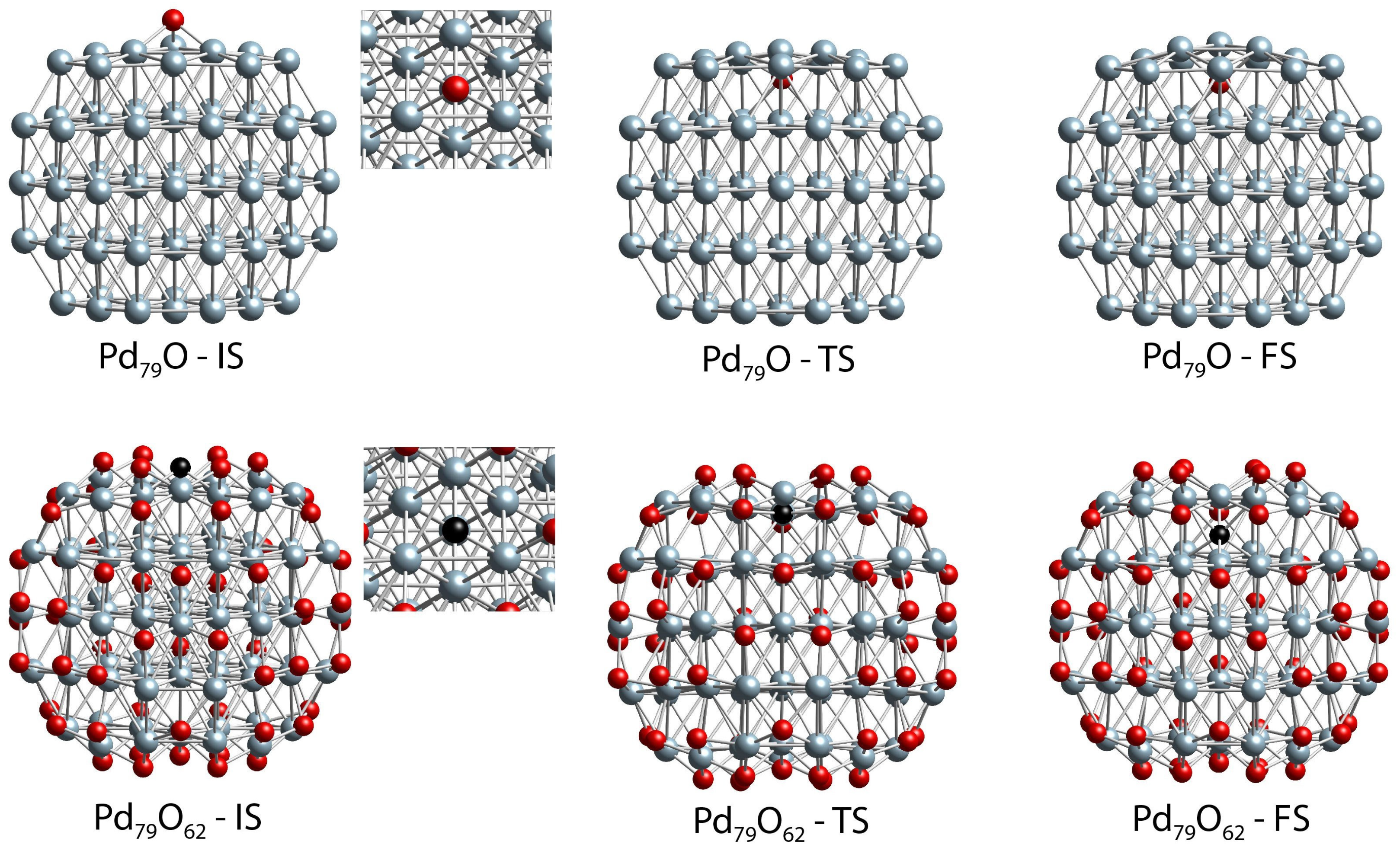
2.4. DFT Modeling of the Interaction of O Species with Pd(111) Surface and Pd79 Nanoparticle Models
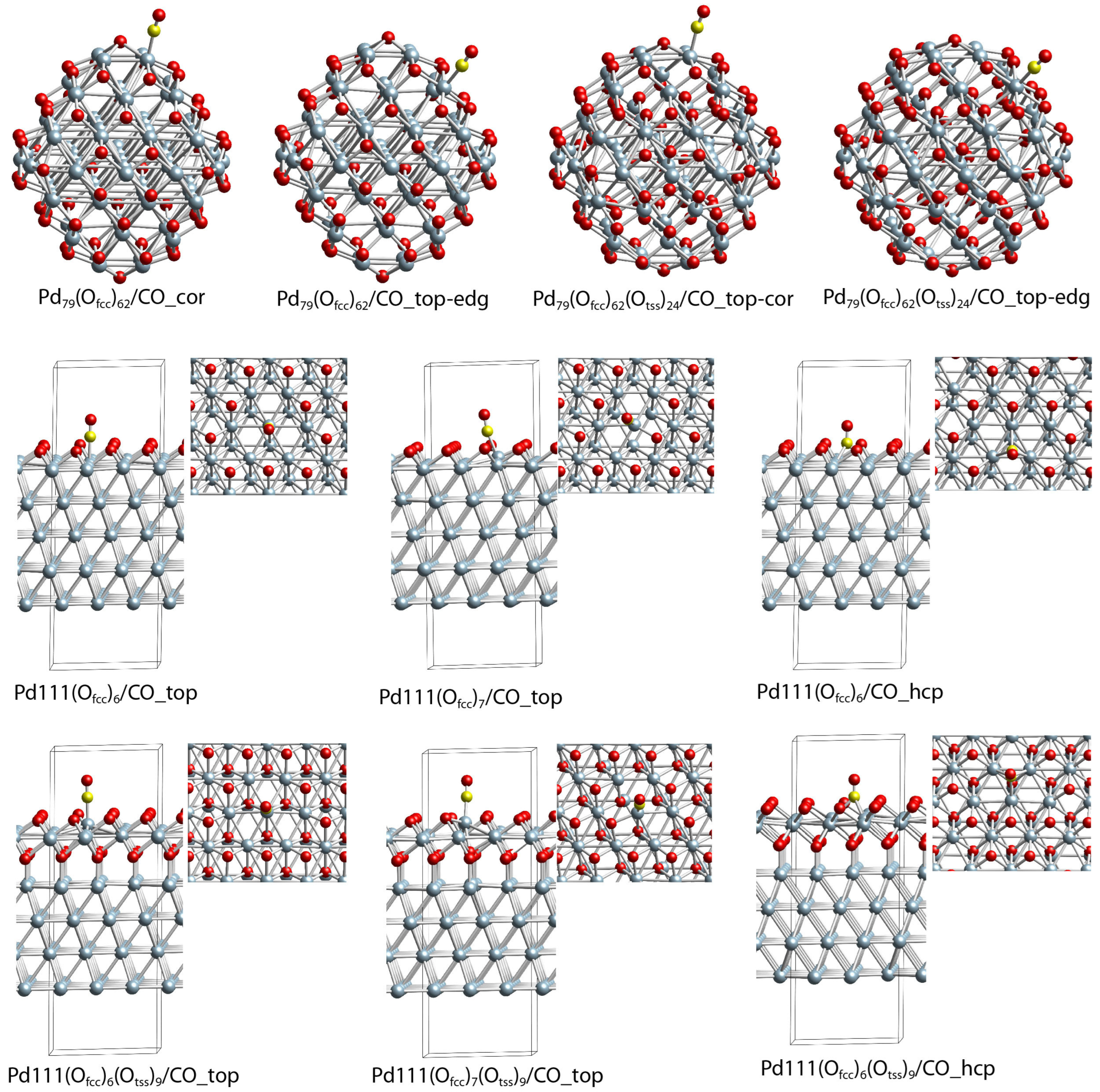
2.5. DFT Modeling Adsorption of CO Species on Oxidized Pd(111) and Pd Nanoparticle
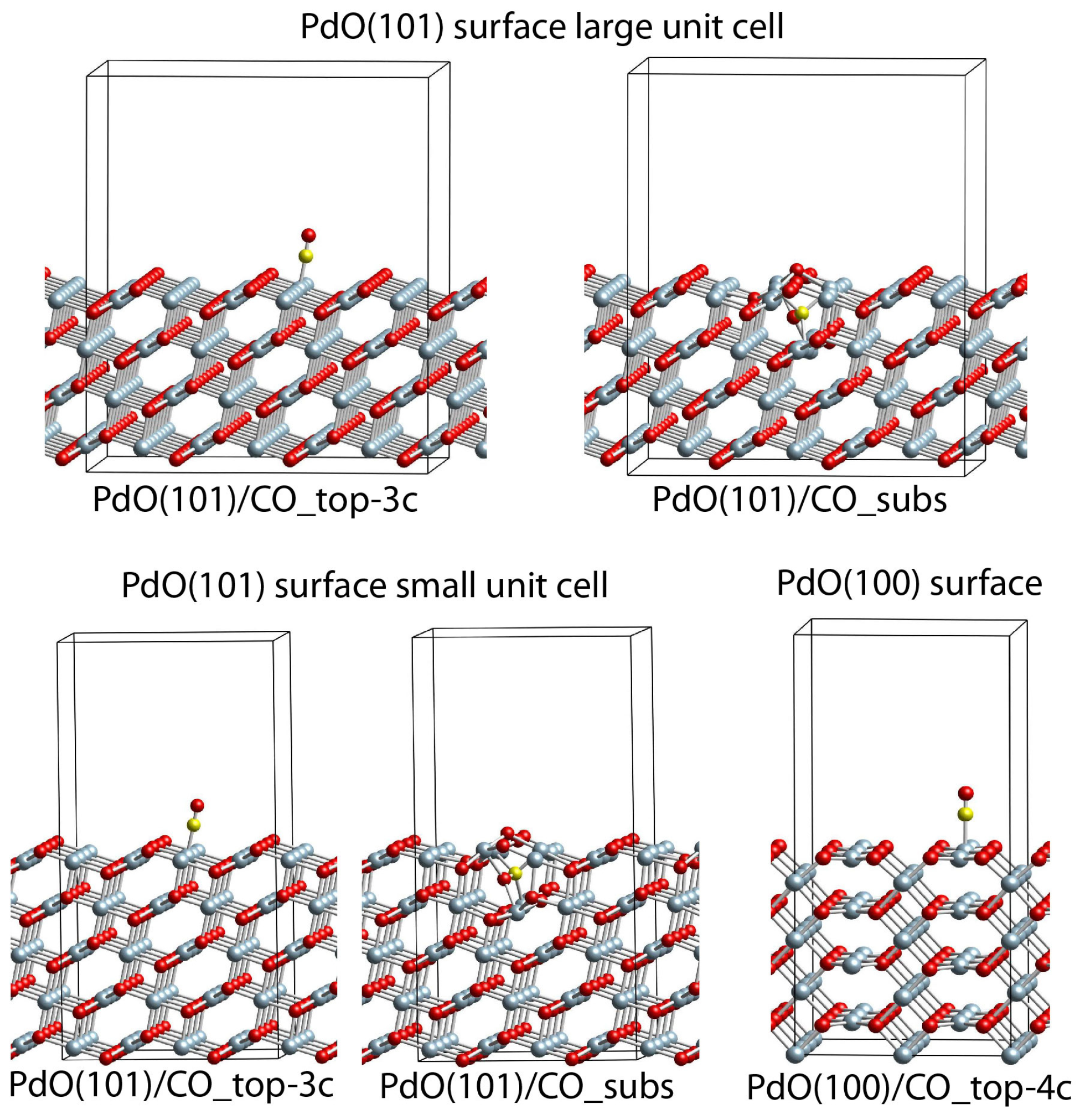
2.6. DFT Modeling Adsorption of CO Species on PdO(101) and PdO(100) Slab Models
2.7. Assignment of the Experimental C-O Vibrational Frequencies
3. Materials and Methods
3.1. Catalyst Preparation
3.2. CH4 Oxidation Reaction Tests and CO Titration
3.3. Catalyst Characterization
3.4. Computational Details and Models
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Oh, D.G.; Lee, J.; Kim, E.; Jang, E.J.; Kim, J.M.; Kwak, J.H. Pd/SiO2 as an active and durable CH4 oxidation catalyst for vehicle applications. J. Ind. Eng. Chem. 2021, 99, 90–97. [Google Scholar] [CrossRef]
- Stasinska, B.; Machocki, A.; Antoniak, K.; Rotko, M.; Figueiredo, J.L.; Gonçalves, F. Importance of palladium dispersion in Pd/Al2O3 catalysts for complete oxidation of humid low-methane–air mixtures. Catal. Today 2008, 137, 329–334. [Google Scholar] [CrossRef]
- Persson, K.; Pfefferle, L.D.; Schwartz, W.; Ersson, A.; Järås, S.G. Stability of palladium-based catalysts during catalytic combustion of methane: The influence of water. Appl. Catal. B Environ. 2007, 74, 242–250. [Google Scholar] [CrossRef]
- Kinnunen, N.M.; Hirvi, J.T.; Suvanto, M.; Pakkanen, T.A. Methane combustion activity of Pd–PdOx–Pt/Al2O3 catalyst: The role of platinum promoter. J. Mol. Catal. A Chem. 2012, 356, 20–28. [Google Scholar] [CrossRef]
- Castellazzi, P.; Groppi, G.; Forzatti, P.; Finocchio, E.; Busca, G. Activation process of Pd/Al2O3 catalysts for CH4 combustion by reduction/oxidation cycles in CH4-containing atmosphere. J. Catal. 2010, 275, 218–227. [Google Scholar] [CrossRef]
- Mihai, O.; Smedler, G.; Nylén, U.; Olofsson, M.; Olsson, L. The effect of water on methane oxidation over Pd/Al2O3 under lean, stoichiometric and rich conditions. Catal. Sci. Technol. 2017, 7, 3084–3096. [Google Scholar] [CrossRef]
- Lin, J.; Chen, X.; Zheng, Y.; Huang, F.; Xiao, Y.; Zheng, Y.; Jiang, L. Facile construction of ultrastable alumina anchored palladium catalysts via a designed one pot strategy for enhanced methane oxidation. Catal. Sci. Technol. 2020, 10, 4612–4623. [Google Scholar] [CrossRef]
- Lin, J.; Zhao, L.; Zheng, Y.; Xiao, Y.; Yu, G.; Zheng, Y.; Chen, W.; Jiang, L. Facile Strategy to Extend Stability of Simple Component-Alumina-Supported Palladium Catalysts for Efficient Methane Combustion. ACS Appl. Mater. Interfaces 2020, 12, 56095–56107. [Google Scholar] [CrossRef]
- Müller, C.A.; Maciejewski, M.; Koeppel, R.A.; Tschan, R.; Baiker, A. Role of Lattice Oxygen in the Combustion of Methane over PdO/ZrO2: Combined Pulse TG/DTA and MS Study with 18O-Labeled Catalyst. J. Phys. Chem. 1996, 100, 20006–20014. [Google Scholar] [CrossRef]
- Fujimoto, K.-i.; Ribeiro, F.H.; Avalos-Borja, M.; Iglesia, E. Structure and Reactivity of PdOx/ZrO2 Catalysts for Methane Oxidation at Low Temperatures. J. Catal. 1998, 179, 431–442. [Google Scholar] [CrossRef] [Green Version]
- Au-Yeung, J.; Chen, K.; Bell, A.T.; Iglesia, E. Isotopic Studies of Methane Oxidation Pathways on PdO Catalysts. J. Catal. 1999, 188, 132–139. [Google Scholar] [CrossRef] [Green Version]
- Jang, E.J.; Lee, J.; Oh, D.G.; Kwak, J.H. CH4 Oxidation Activity in Pd and Pt–Pd Bimetallic Catalysts: Correlation with Surface PdOx Quantified from the DRIFTS Study. ACS Catal. 2021, 11, 5894–5905. [Google Scholar] [CrossRef]
- Lyubovsky, M.; Pfefferle, L. Complete methane oxidation over Pd catalyst supported on α-alumina. Influence of temperature and oxygen pressure on the catalyst activity. Catal. Today 1999, 47, 29–44. [Google Scholar] [CrossRef]
- Xu, J.; Ouyang, L.; Mao, W.; Yang, X.-J.; Xu, X.-C.; Su, J.-J.; Zhuang, T.-Z.; Li, H.; Han, Y.-F. Operando and Kinetic Study of Low-Temperature, Lean-Burn Methane Combustion over a Pd/γ-Al2O3 Catalyst. ACS Catal. 2012, 2, 261–269. [Google Scholar] [CrossRef]
- Hellman, A.; Resta, A.; Martin, N.M.; Gustafson, J.; Trinchero, A.; Carlsson, P.A.; Balmes, O.; Felici, R.; van Rijn, R.; Frenken, J.W.; et al. The Active Phase of Palladium during Methane Oxidation. J. Phys. Chem. Lett. 2012, 3, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Zorn, K.; Giorgio, S.; Halwax, E.; Henry, C.R.; Grönbeck, H.; Rupprechter, G. CO Oxidation on Technological Pd−Al2O3 Catalysts: Oxidation State and Activity. J. Phys. Chem. C 2011, 115, 1103–1111. [Google Scholar] [CrossRef]
- Mehar, V.; Kim, M.; Shipilin, M.; Van den Bossche, M.; Gustafson, J.; Merte, L.R.; Hejral, U.; Grönbeck, H.; Lundgren, E.; Asthagiri, A.; et al. Understanding the Intrinsic Surface Reactivity of Single-Layer and Multilayer PdO(101) on Pd(100). ACS Catal. 2018, 8, 8553–8567. [Google Scholar] [CrossRef]
- Datye, A.K.; Bravo, J.; Nelson, T.R.; Atanasova, P.; Lyubovsky, M.; Pfefferle, L. Catalyst microstructure and methane oxidation reactivity during the Pd↔PdO transformation on alumina supports. Appl. Catal. A Gen. 2000, 198, 179–196. [Google Scholar] [CrossRef]
- Xiong, H.; Lester, K.; Ressler, T.; Schlögl, R.; Allard, L.F.; Datye, A.K. Metastable Pd ↔ PdO Structures During High Temperature Methane Oxidation. Catal. Lett. 2017, 147, 1095–1103. [Google Scholar] [CrossRef]
- Farrauto, R.J.; Hobson, M.C.; Kennelly, T.; Waterman, E.M. Catalytic chemistry of supported palladium for combustion of methane. Appl. Catal. A Gen. 1992, 81, 227–237. [Google Scholar] [CrossRef]
- Farrauto, R.J.; Lampert, J.K.; Hobson, M.C.; Waterman, E.M. Thermal decomposition and reformation of PdO catalysts; support effects. Appl. Catal. B Environ. 1995, 6, 263–270. [Google Scholar] [CrossRef]
- Mouaddib, N.; Feumijantou, C.; Garbowski, E.; Primet, M. Catalytic oxidation of methane over palladium supported on alumina: Influence of the oxygen-to-methane ratio. Appl. Catal. A-Gen. 1992, 87, 129–144. [Google Scholar] [CrossRef]
- Gelin, P.; Primet, M. Complete oxidation of methane at low temperature over noble metal based catalysts: A review. Appl. Catal. B-Environ. 2002, 39, 1–37. [Google Scholar] [CrossRef]
- Eguchi, K.; Arai, H. Recent advances in high temperature catalytic combustion. Catal. Today 1996, 29, 379–386. [Google Scholar] [CrossRef]
- Choudhary, T.V.; Banerjee, S.; Choudhary, V.R. Catalysts for combustion of methane and lower alkanes. Appl. Catal. A-Gen. 2002, 234, 1–23. [Google Scholar] [CrossRef]
- Cullis, C.F.; Willatt, B.M. Oxidation of methane over supported precious metal catalysts. J. Catal. 1983, 83, 267–285. [Google Scholar] [CrossRef]
- Eguchi, K.; Arai, H. Low temperature oxidation of methane over Pd-based catalysts—Effect of support oxide on the combustion activity. Appl. Catal. A-Gen. 2001, 222, 359–367. [Google Scholar] [CrossRef]
- Ciuparu, D.; Lyubovsky, M.R.; Altman, E.; Pfefferle, L.D.; Datye, A. Catalytic combustion of methane over palladium-based catalysts. Catal. Rev. 2002, 44, 593–649. [Google Scholar] [CrossRef]
- Lampert, J.K.; Kazi, M.S.; Farrauto, R.J. Palladium catalyst performance for methane emissions abatement from lean burn natural gas vehicles. Appl. Catal. B-Environ. 1997, 14, 211–223. [Google Scholar] [CrossRef]
- Sekizawa, K.; Widjaja, H.; Maeda, S.; Ozawa, Y.; Eguchi, K. Low temperature oxidation of methane over Pd catalyst supported on metal oxides. Catal. Today 2000, 59, 69–74. [Google Scholar] [CrossRef]
- Kovarik, L.; Jaegers, N.; Szanyi, J.; Derewinski, M.; Wang, Y.; Khivantsev, K. PdO self-assembly on zeolite SSZ-13 with rows of O3Al(IV)OH selectively incorporated in PdO(101) facets for moisture-resistant methane oxidation. ChemRxiv 2021. [Google Scholar] [CrossRef]
- Kan, H.H.; Colmyer, R.J.; Asthagiri, A.; Weaver, J.F. Adsorption of Water on a PdO(101) Thin Film: Evidence of an Adsorbed HO−H2O Complex. J. Phys. Chem. C 2009, 113, 1495–1506. [Google Scholar] [CrossRef]
- Lee, J.; Young Kim, M.; Hong Jeon, J.; Lee, D.H.; Rao, K.N.; Oh, D.G.; Jeong Jang, E.; Kim, E.; Na, S.C.; Han, H.S.; et al. Effect of Pt pre-sintering on the durability of PtPd/Al2O3 catalysts for CH4 oxidation. Appl. Catal. B Environ. 2020, 260, 118098. [Google Scholar] [CrossRef]
- Gun Oh, D.; Aleksandrov, H.A.; Kim, H.; Koleva, I.Z.; Khivantsev, K.; Vayssilov, G.N.; Hun Kwak, J. Key Role of a-Top CO on Terrace Sites of Metallic Pd Clusters for CO Oxidation. Chem.–A Eur. J. 2022, 28, e202200684. [Google Scholar] [CrossRef]
- Hadjiivanov, K.I.; Vayssilov, G.N. Characterization of oxide surfaces and zeolites by carbon monoxide as an IR probe molecule. In Advances in Catalysis; Academic Press: Cambridge, MA, USA, 2002; Volume 47, pp. 307–511. [Google Scholar] [CrossRef]
- Martin, N.M.; Van den Bossche, M.; Grönbeck, H.; Hakanoglu, C.; Zhang, F.; Li, T.; Gustafson, J.; Weaver, J.F.; Lundgren, E. CO Adsorption on Clean and Oxidized Pd(111). J. Phys. Chem. C 2014, 118, 1118–1128. [Google Scholar] [CrossRef]
- Kresse, G.; Hafner, J. Ab initio molecular-dynamics simulation of the liquid-metal--amorphous-semiconductor transition in germanium. Phys. Rev. B 1994, 49, 14251–14269. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef]
- Blöchl, P.E. Projector augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 1999, 59, 1758–1775. [Google Scholar] [CrossRef]
- Monkhorst, H.J.; Pack, J.D. Special points for Brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Methfessel, M.; Paxton, A.T. High-precision sampling for Brillouin-zone integration in metals. Phys. Rev. B 1989, 40, 3616–3621. [Google Scholar] [CrossRef] [PubMed] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure | E≠ | ΔE | Pd-O | Pd-Pd |
|---|---|---|---|---|
| Pd79O-IS | 2.28 | 2.26 | 199 a;199 a;199 a | 286;287;287 |
| Pd79O-TS | 201 a;202 a;202 a/232 b;233 b;237 b | 308;310;312 | ||
| Pd79O-FS | 210 a;210 a;211 a/222 b;223 b;223 b | 288;288;288 | ||
| Pd79O62-IS | 1.17 | 0.31 | 198 a;198 a;198 a | 298;298;298 |
| Pd79O62-TS | 195 a;196 a;196 a | 338;339;340 | ||
| Pd79O62-FS | 218 a;218 a;219 a/216 b;216 b;216 b | 277;277;277 |
| Structure | BE a | CN b | r(Pd-C) | ν(C-O) | Δr(C-O) c |
|---|---|---|---|---|---|
| Oxidized palladium surface | |||||
| Pd111(Ofcc)7/CO_top | −0.17 | 9 | 194 | 2085 | 0.5 |
| Pd111(Ofcc)6/CO_hcp | −0.92 | 9;9;9 | 211;211;211 | 1873 | 3.4 |
| Pd111(Ofcc)6/CO_top | −0.99 | 9 | 192 | 2072 | 0.8 |
| Pd111(Ofcc)7(Otss)9/CO_top | −0.26 | 9 | 189 | 2090 | 0.7 |
| Pd111(Ofcc)6(Otss)9/CO_hcp | −1.02 | 9;9;9 | 214;214;215 | 1889 | 3.2 |
| Pd111(Ofcc)6(Otss)9/CO_top | −1.65 | 9 | 187 | 2083 | 0.9 |
| Oxidized palladium nanoparticle | |||||
| Pd79(Ofcc)62/CO_top-cor | −0.49 | 6 | 200 | 2088 | 0.4 |
| Pd79(Ofcc)62/CO_top-edg | 0.04 | 7 | 203 | 2121 | −0.1 |
| Pd79(Ofcc)59/CO_top-cen | −1.48 | 9 | 188 | 2051 | 1.2 |
| Pd79(Ofcc)62(Otss)24/CO_top-cor | −0.37 | 6 | 201 | 2095 | 0.2 |
| Pd79(Ofcc)62(Otss)24/CO_top-edg | −0.63 | 7 | 194 | 2120 | 0.0 |
| PdO(101) surface large unit cell | |||||
| PdO(101)/CO_top-3c | −1.49 | 3 | 189 | 2090 | 0.7 |
| PdO(101)/CO_subs d | 3.18 | 2;4;4 | 203;214;218 | 1540 | 7.2 |
| PdO(101) surface small unit cell | |||||
| PdO(101)/CO_top-3c | −1.56 | 3 | 189 | 2084 | 0.8 |
| PdO(101)/CO_subs e | 3.13 | 2;4;4 | 201;214;218 | 1499 | 8.1 |
| PdO(100) surface | |||||
| PdO(100)/CO_top-4c | −0.04 | 4 | 203 | 2131 | −0.1 |
| Structures | CN a | νcalc | νexp |
|---|---|---|---|
| On-top slab: Pd111/CO_top | 2036 | 2087 (51) | |
| On-top terrace: Pd79/CO_top-cen b | 2023 | 2087 (64) | |
| On-top edge: Pd79/CO_top-edg b | 2015 | 2068 (53) | |
| On-top corner: Pd79/CO_top-cor b | 2013 | 2068 (55) | |
| Pd111(Ofcc)6(Otss)9/CO_top | 3 | 2083 | 2143 (60) |
| Pd111(Ofcc)7/CO_top | 3 | 2085 | 2143 (58) |
| Pd111(Ofcc)7(Otss)9/CO_top | 3 | 2090 | 2143 (53) |
| Pd79(Ofcc)62/CO_top-cor | 3 | 2088 | 2143 (55) |
| PdO(101)/CO_top-3c | 3 | 2090 | 2143 (53) |
| Pd79(Ofcc)62(Otss)24/CO_top-cor | 3 | 2095 | 2143 (48) |
| Pd79(Ofcc)62/CO_top-edg | 4 | 2121 | 2160 (39) |
| Pd79(Ofcc)62(Otss)24/CO_top-edg | 4 | 2120 | 2160 (40) |
| PdO(100)/CO_top-4c | 4 | 2131 | 2160 (29) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oh, D.G.; Aleksandrov, H.A.; Kim, H.; Koleva, I.Z.; Khivantsev, K.; Vayssilov, G.N.; Kwak, J.H. Understanding of Active Sites and Interconversion of Pd and PdO during CH4 Oxidation. Molecules 2023, 28, 1957. https://doi.org/10.3390/molecules28041957
Oh DG, Aleksandrov HA, Kim H, Koleva IZ, Khivantsev K, Vayssilov GN, Kwak JH. Understanding of Active Sites and Interconversion of Pd and PdO during CH4 Oxidation. Molecules. 2023; 28(4):1957. https://doi.org/10.3390/molecules28041957
Chicago/Turabian StyleOh, Dong Gun, Hristiyan A. Aleksandrov, Haneul Kim, Iskra Z. Koleva, Konstantin Khivantsev, Georgi N. Vayssilov, and Ja Hun Kwak. 2023. "Understanding of Active Sites and Interconversion of Pd and PdO during CH4 Oxidation" Molecules 28, no. 4: 1957. https://doi.org/10.3390/molecules28041957
APA StyleOh, D. G., Aleksandrov, H. A., Kim, H., Koleva, I. Z., Khivantsev, K., Vayssilov, G. N., & Kwak, J. H. (2023). Understanding of Active Sites and Interconversion of Pd and PdO during CH4 Oxidation. Molecules, 28(4), 1957. https://doi.org/10.3390/molecules28041957