


Multicomponent Domino Cyclization of Ethyl Trifluoropyruvate with Methyl Ketones and Amino Alcohols as A New Way to γ-Lactam Annulated Oxazacycles
 , ,
, ,
Abstract
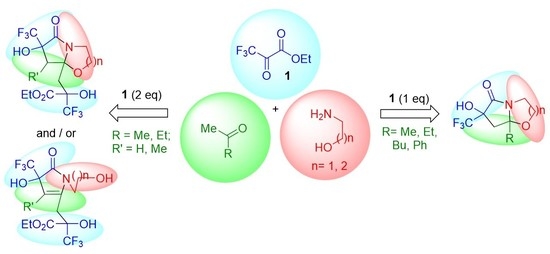
:
1. Introduction
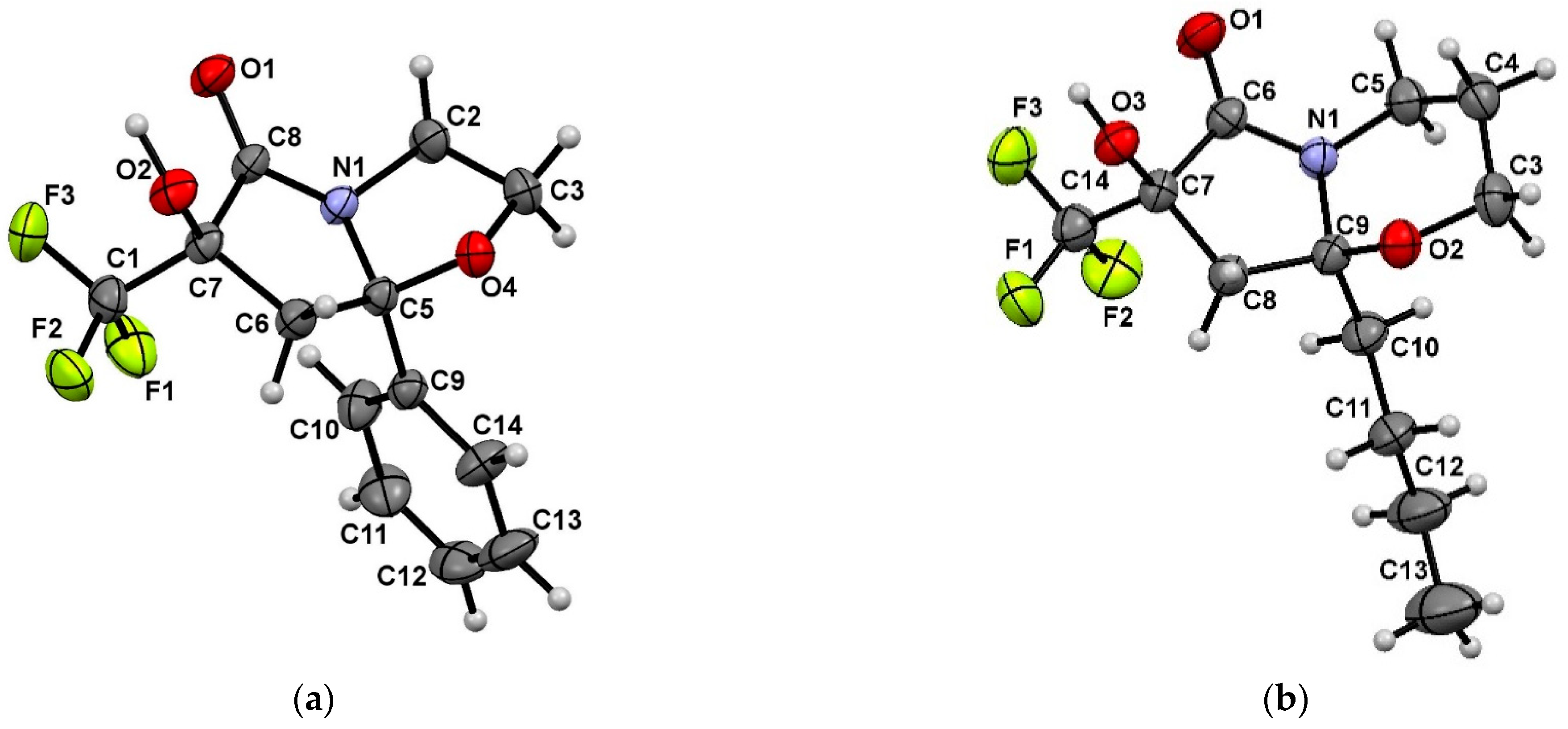
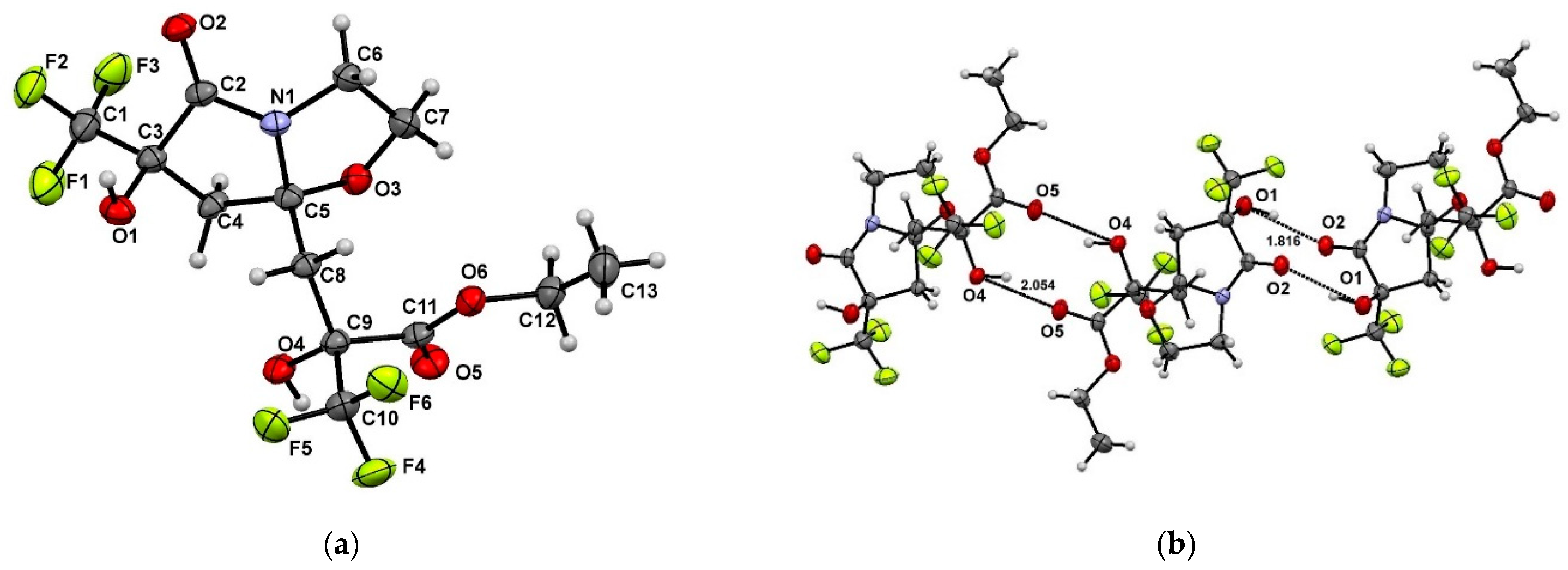
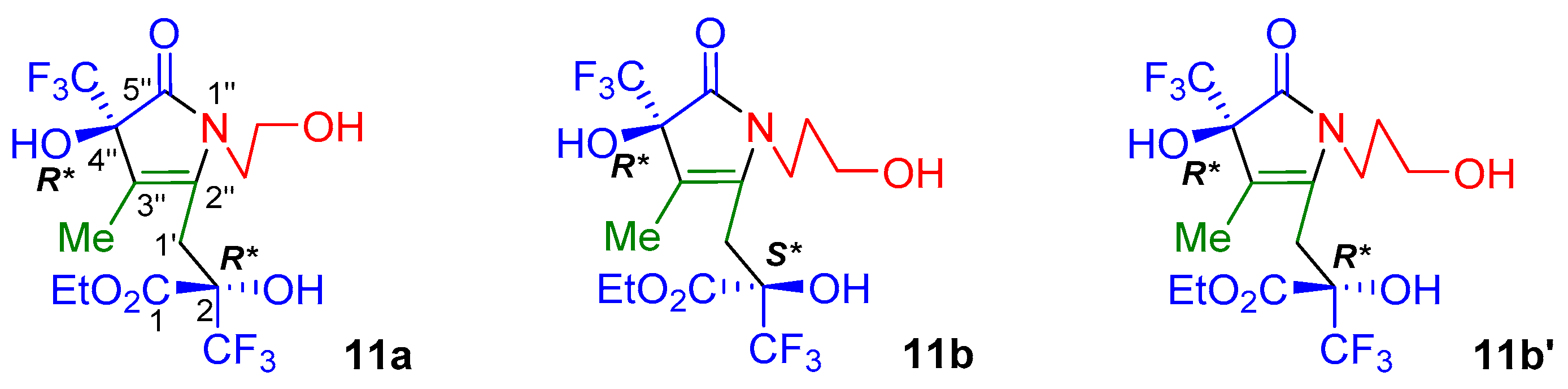
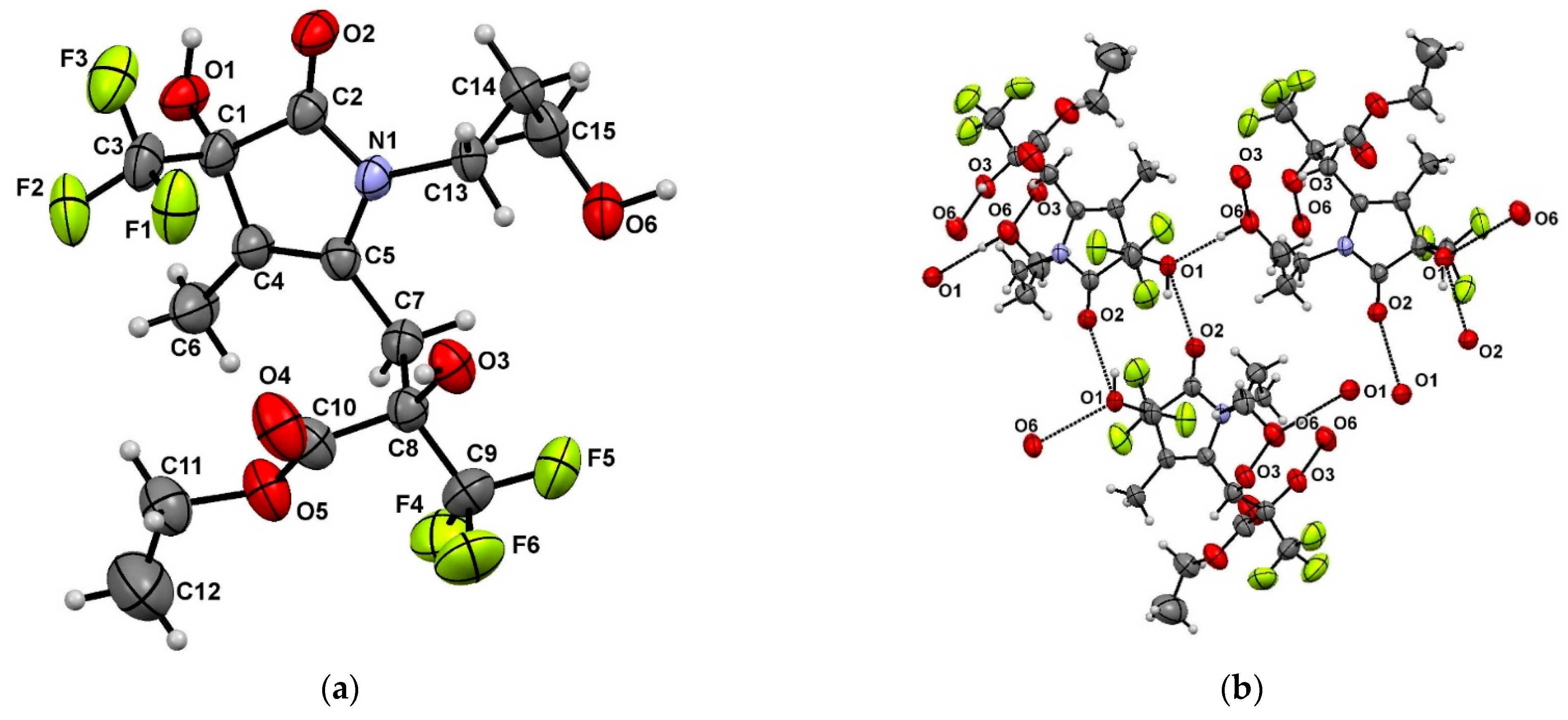
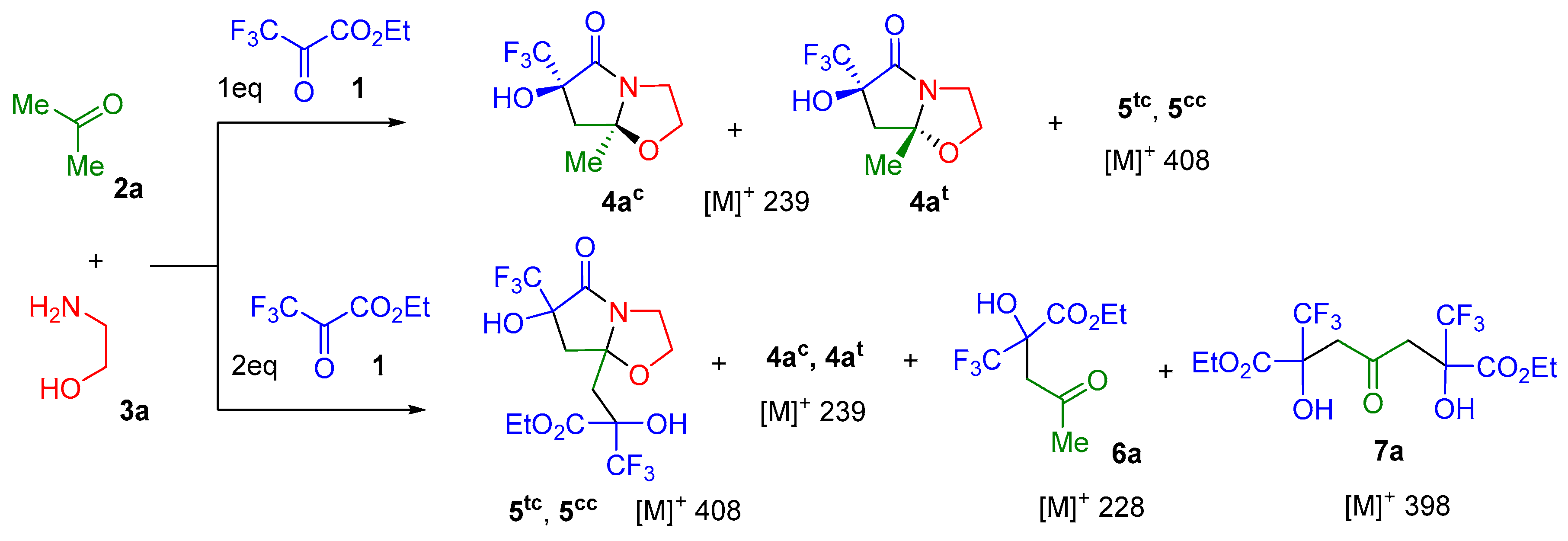
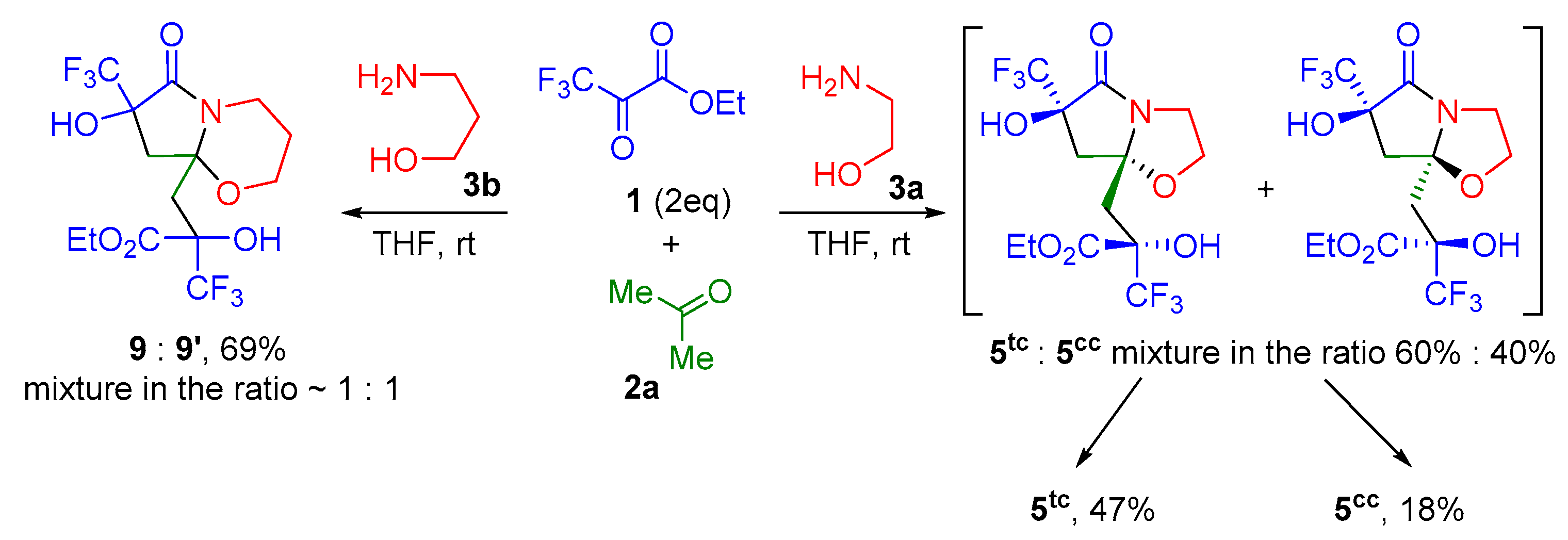
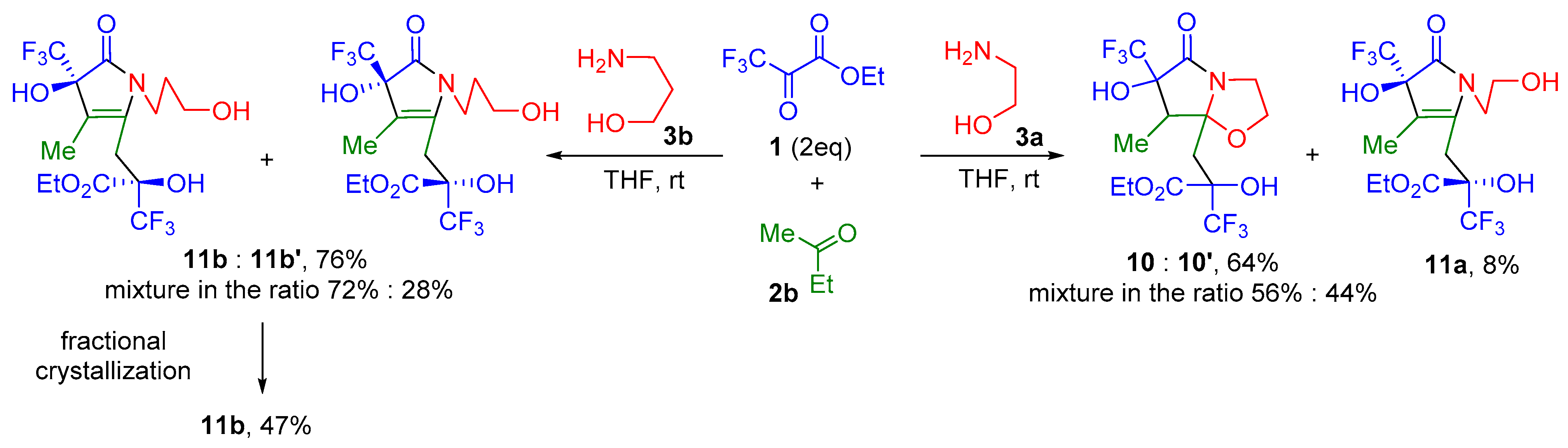
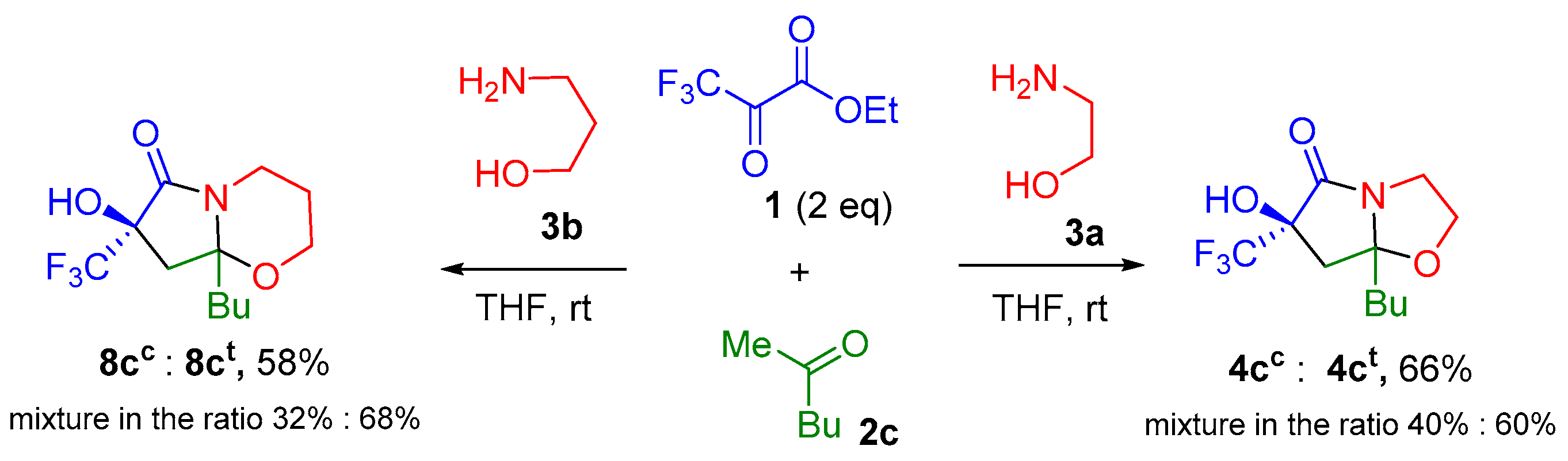
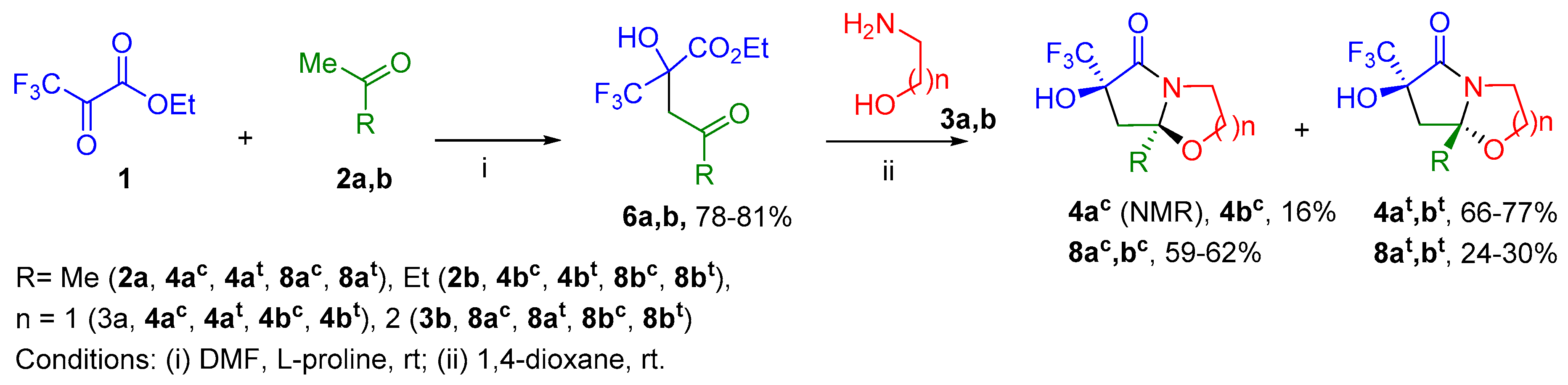
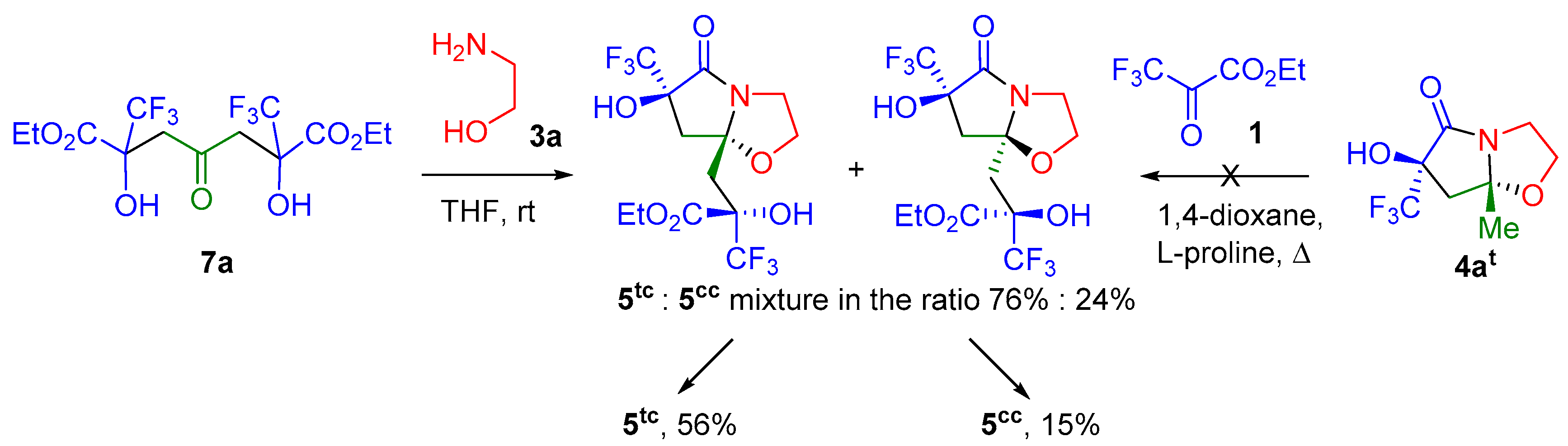
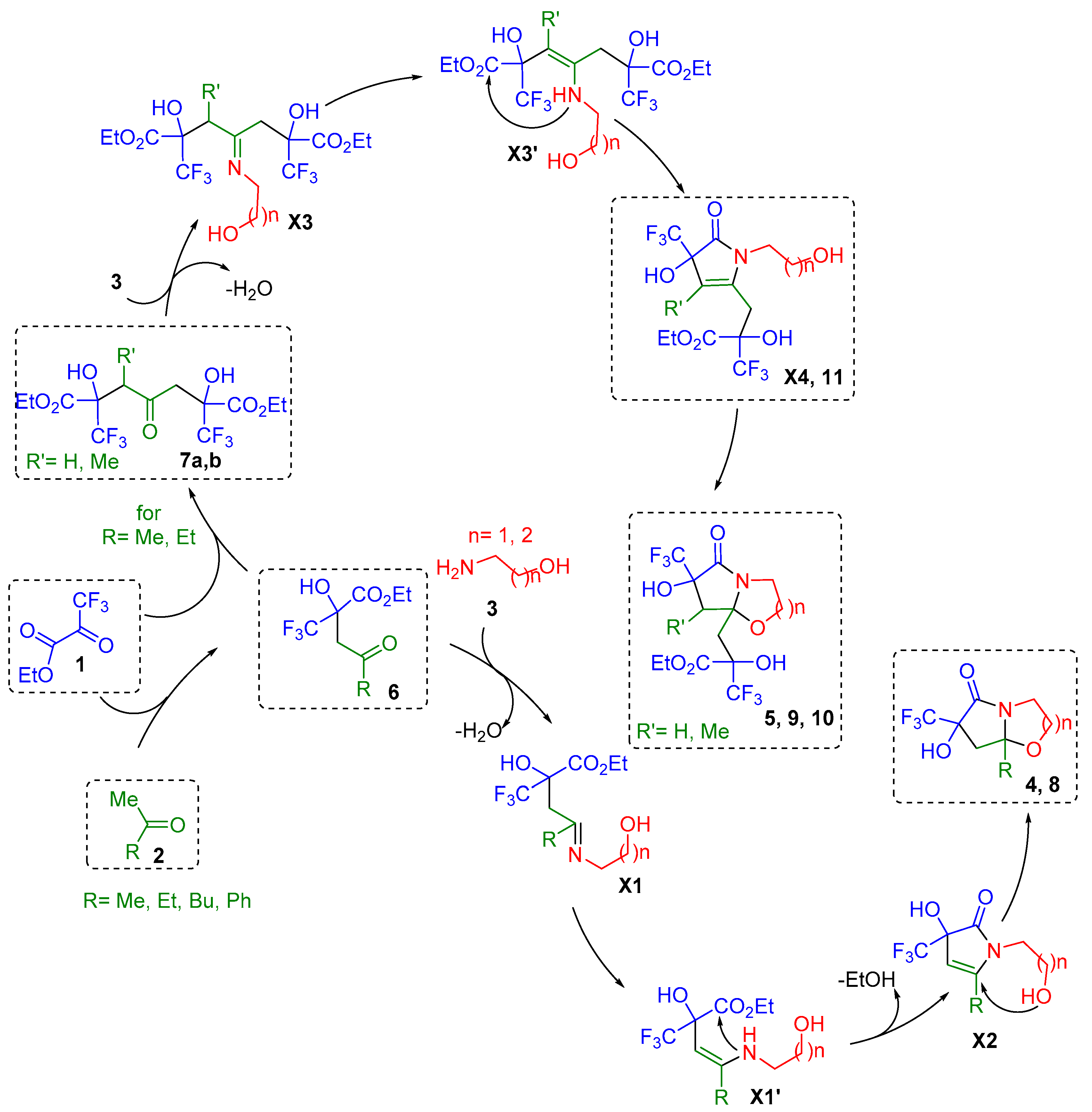
2. Results
3. Material and Methods
3.1. Material
3.2. Methods
3.3. General Procedures
3.4. Spectral Data
3.5. XRD Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Caruano, J.; Muccioli, G.G.; Robiette, R. Biologically active γ-lactams: Synthesis and natural sources. Org. Biomol. Chem. 2016, 14, 10134–10156. [Google Scholar] [CrossRef]
- Groaning, M.D.; Meyers, A.I. Concise synthesis of indolizidines: Total synthesis of (−)-coniceine. Chem. Commun. 2000, 1027–1028. [Google Scholar] [CrossRef]
- Fenteany, G.; Standaert, R.F.; Lane, W.S.; Choi, S.; Corey, E.J.; Schreiber, S.L. Inhibition of proteasome activities and subunit-specific amino-terminal threonine modification by Lactacystin. Science 1995, 268, 726–731. [Google Scholar] [CrossRef]
- Chu, S.; Liu, S.; Duan, W.; Cheng, Y.; Jiang, X.; Zhu, C.; Tang, K.; Wang, R.; Xu, L.; Wang, X.; et al. The anti-dementia drug candidate, (−)-clausenamide, improves memory impairment through its multi-target effect. Pharmacol. Ther. 2016, 162, 179–187. [Google Scholar] [CrossRef]
- Yost, C.S. A new look at the Respiratory Stimulant Doxapram. CNS Drug Rev. 2006, 12, 236–249. [Google Scholar] [CrossRef]
- Glauser, T.A.; Cnaan, A.; Shinnar, S.; Hirtz, D.G.; Dlugos, D.; Masur, D.; Clark, P.O.; Capparelli, E.V.; Adamson, P.C. Ethosuximide, valproic acid, and lamotrigine in childhood absence epilepsy. New Engl. J. Med. 2010, 362, 790–799. [Google Scholar] [CrossRef] [Green Version]
- Shorvon, S.D. Piracetam. In the Treatment of Epilepsy, 3rd ed.; Shorvon, S., Perucca, E., Engel, J., Eds.; Wiley-Blackwell: Oxford, UK, 2009; pp. 619–625. [Google Scholar] [CrossRef]
- Pinza, M.; Farina, C.; Cerri, A.; Pfeiffer, U.; Riccaboni, M.T.; Banfi, S.; Biagetti, R.; Pozzi, O.; Magnani, M.; Dorigotti, L. Synthesis and pharmacological activity of a series of dihydro-1H-pyrrolo[1,2-a]imidazole-2,5(3H,6H)-diones, a novel class of potent cognition enhancers. J. Med. Chem. 1993, 36, 4214–4220. [Google Scholar] [CrossRef]
- Boyd, D.B.; Elzey, T.K.; Hatfield, L.D.; Kinnick, M.D.; Morin, J.M. γ-Lactam analogues of the penems. Tetrahedron Lett. 1986, 27, 3453–3456. [Google Scholar] [CrossRef]
- Baldwin, J.E.; Adlington, R.M.; Jones, R.H.; Schofield, C.J.; Zarocostas, C.; Greengrass, C.W. γ-Lactam analogues of carbapenicillanic acids. J. Chem. Soc. Chem. Commun. 1985, 194–196. [Google Scholar] [CrossRef]
- Groaning, M.D.; Meyers, A. Chiral non-racemic bicyclic lactams. Auxiliary-based asymmetric reactions. Tetrahedron 2000, 56, 9843–9873. [Google Scholar] [CrossRef]
- Romo, D.; Meyers, A. Chiral non-racemic bicyclic lactams. Vehicles for the construction of natural and unnatural products containing quaternary carbon centers. Tetrahedron 1991, 47, 9503–9569. [Google Scholar] [CrossRef]
- Meyers, A.I.; Harre, M.; Garland, R. Asymmetric synthesis of quaternary carbon centers. J. Am. Chem. Soc. 1984, 106, 1146–1148. [Google Scholar] [CrossRef]
- Meyers, A.I.; Lefker, B.A. Chiral bicyclic lactams for asymmetric synthesis of quaternary carbons. The total synthesis of (-)-.alpha.-cuparenone. J. Org. Chem. 1986, 51, 1541–1544. [Google Scholar] [CrossRef]
- Malaquin, S.; Jida, M.; Courtin, J.; Laconde, G.; Willand, N.; Deprez, B.; Deprez-Poulain, R. Water-based conditions for the microscale parallel synthesis of bicyclic lactams. Tetrahedron Lett. 2013, 54, 562–567. [Google Scholar] [CrossRef]
- Le Goff, R.; Martel, A.; Sanselme, M.; Lawson, A.M.; Daïch, A.; Comesse, S. Simple access to highly functional bicyclic γ- and δ-lactams: Origins of chirality transfer to contiguous tertiary/quaternary stereocenters assessed by DFT. Chem. A Eur. J. 2014, 21, 2966–2979. [Google Scholar] [CrossRef]
- Comesse, S.; Martel, A.; Daïch, A. Domino process optimized via ab initio study for an alternative access to bicyclic lactams. Org. Lett. 2011, 13, 4004–4007. [Google Scholar] [CrossRef]
- Pilard, J.-F.; Klein, B.; Texier-Boullet, F.; Hamelin, J. Fast synthesis of heterobicycles containing a bridgehead nitrogen atom in dry media under microwave irradiation. Synlett 1992, 1992, 219–220. [Google Scholar] [CrossRef]
- Wedler, C.; Schick, H.; Scharfenberg-Pfeiffer, D.; Reck, G. Reactions of 4-oxoalkanoic acids, 4. synthesis of (±)-6-alkyl-5-oxa-1-azabicyclo[4.3.0]nonan-9-ones, (±)-5-alkyl-4-oxa-1-azabicyclo[3.3.0]octan-8-ones, and substituted 1,6-dioxa-3,8-diazacyclodecanes by reaction of ethyl 4-oxoalkanoates with 3-aminopropanol and 2-aminoethanol. Eur. J. Org. Chem. 1992, 1992, 29–32. [Google Scholar] [CrossRef]
- Vainiotalo, P.; Savolainen, P.-L.; Ahlgrén, M.; Mälkönen, P.J.; Vepsäläinen, J. Identification of the condensation products of 1,2- and 1,3-amino alcohols with keto esters by NMR spectroscopy, mass spectrometry and X-ray crystallography studies. Open or ring forms? J. Chem. Soc. Perkin Trans. 2 1991, 735–741. [Google Scholar] [CrossRef]
- Meyers, A.I.; Wallace, R.H. Some observations on the validity and generality of the "Cieplak stereoelectronic effect". J. Org. Chem. 1989, 54, 2509–2510. [Google Scholar] [CrossRef]
- Hao, J.; Hou, M.; Zhuang, H.; Wang, J.; Wan, W.; Jiang, H. A Process for Preparing (3S)-3-Benzyl-7a-(Trifluoromethyl)Tetrahydropyrrolo[2,1-b]Oxazol-5(6H)-One. Patent CN101531668 A China, 2009.
- Hao, J.; Hou, M.; Zhuang, H.; Wang, J.; Wan, W.; Jiang, H. Preparation of (3R)-3-Benzyl-7a-(Difluoromethyl)Tetrahydropyrrolo[2,1-b]Oxazol-5(6H)-One. Patent CN101544658 A China, 2009.
- Li, B.; Guinness, S.M.; Hoagland, S.; Fichtner, M.; Kim, H.; Li, S.; Maguire, R.J.; McWilliams, J.C.; Mustakis, J.; Raggon, J.; et al. Continuous production of anhydrous tert-butyl hydroperoxide in nonane using membrane pervaporation and its application in flow oxidation of a γ-butyrolactam. Org. Process Res. Dev. 2018, 22, 707–720. [Google Scholar] [CrossRef]
- Bhatt, J.D.; Patel, T.S.; Chudasama, C.J.; Patel, K.D. Microwave-assisted synthesis of novel pyrazole clubbed polyhydroquinolines in an ionic-liquid and their biological perspective. ChemistrySelect 2018, 3, 3632–3640. [Google Scholar] [CrossRef]
- Wu, Y.; Wang, Y.; He, M.; Tao, X.; Li, J.; Shan, D.; Lv, L. Progress of trifluoroacetoacetate in the synthesis of agrochemicals and medicines with fluorine. Mini-Rev. Org. Chem. 2017, 14, 350–356. [Google Scholar] [CrossRef]
- O’Hagan, D. Understanding organofluorine chemistry. An introduction to the C–F bond. Chem. Soc. Rev. 2007, 37, 308–319. [Google Scholar] [CrossRef]
- Jeffries, B.; Wang, Z.; Graton, J.; Holland, S.D.; Brind, T.; Greenwood, R.D.R.; Le Questel, J.-Y.; Scott, J.S.; Chiarparin, E.; Linclau, B. Reducing the lipophilicity of perfluoroalkyl groups by CF2–F/CF2–Me or CF3/CH3 exchange. J. Med. Chem. 2018, 61, 10602–10618. [Google Scholar] [CrossRef] [Green Version]
- Giornal, F.; Pazenok, S.; Rodefeld, L.; Lui, N.; Vors, J.-P.; Leroux, F.R. Synthesis of diversely fluorinated pyrazoles as novel active agrochemical ingredients. J. Fluor. Chem. 2013, 152, 2–11. [Google Scholar] [CrossRef]
- Muller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [Green Version]
- Goryaeva, M.V.; Burgart, Y.V.; Kudyakova, Y.S.; Ezhikova, M.A.; Kodess, M.I.; Slepukhin, P.A.; Saloutin, V.I. Three-component synthesis of 7-hydroxy-7-polyfluoroalkylhexahydroimidazo[1,2-a] pyridin-5(1H)-ones. Eur. J. Org. Chem. 2015, 2015, 6306–6314. [Google Scholar] [CrossRef]
- Saloutin, V.I.; Goryaeva, M.V.; Kushch, S.O.; Khudina, O.G.; Ezhikova, M.A.; Kodess, M.I.; Slepukhin, P.A.; Burgart, Y.V. Competitive ways for three-component cyclization of polyfluoroalkyl-3-oxo esters, methyl ketones and amino alcohols. Pure Appl. Chem. 2020, 92, 1265–1275. [Google Scholar] [CrossRef]
- Goryaeva, M.V.; Kushch, S.O.; Khudina, O.G.; Burgart, Y.V.; Ezhikova, M.A.; Kodess, M.I.; Slepukhin, P.A.; Volobueva, A.S.; Slita, A.V.; Esaulkova, I.L.; et al. New multicomponent approach to polyfluoroalkylated pyrido[1,2-a]pyrimidine derivatives and bis-cyclohexenones. J. Fluor. Chem. 2020, 241, 109686. [Google Scholar] [CrossRef]
- Kushch, S.O.; Goryaeva, M.V.; Burgart, Y.V.; Triandafilova, G.A.; Malysheva, K.O.; Krasnykh, O.P.; Gerasimova, N.A.; Evstigneeva, N.P.; Saloutin, V.I. Facile synthesis of 6-organyl-4-(trifluoromethyl)pyridin-2(1H)-ones and their polyfluoroalkyl-containing analogs. Russ. Chem. Bull. 2022, 71, 1687–1700. [Google Scholar] [CrossRef] [PubMed]
- Goryaeva, M.V.; Fefelova, O.; Burgart, Y.V.; Ezhikova, M.; Kodess, M.I.; Slepukhin, P.; Triandafilova, G.; Solodnikov, S.Y.; Krasnykh, O.P.; Saloutin, V.I. A three-component synthesis of trifluoromethylated hexahydropyrrolo[1,2-a]imidazol-5-ones and hexahydropyrrolo[1,2-a]pyrimidin-6-ones. Chem. Heterocycl. Compd. 2022, 58, 421–431. [Google Scholar] [CrossRef]
- Landge, S.M.; Török, B. Highly enantioselective organocatalytic addition of ethyl trifluoropyruvate to ketones with subzero temperature microwave activation. Catal. Lett. 2009, 131, 432–439. [Google Scholar] [CrossRef]
- Goryaeva, M.V.; Kushch, S.O.; Burgart, Y.V.; Ezhikova, M.A.; Kodess, M.I.; Slepukhin, P.A.; Triandafilova, G.A.; Krasnykh, O.P.; Yakovleva, E.I.; Zarubaev, V.V.; et al. New heteroanalogs of tricyclic ascidian alkaloids: Synthesis and biological activity. Org. Biomol. Chem. 2021, 19, 9925–9935. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Palatinus, L.; Chapuis, G. SUPERFLIP—A computer program for the solution of crystal structures by charge flipping in arbitrary dimensions. J. Appl. Crystallogr. 2007, 40, 786–790. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2007, A64, 112–122. [Google Scholar] [CrossRef] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
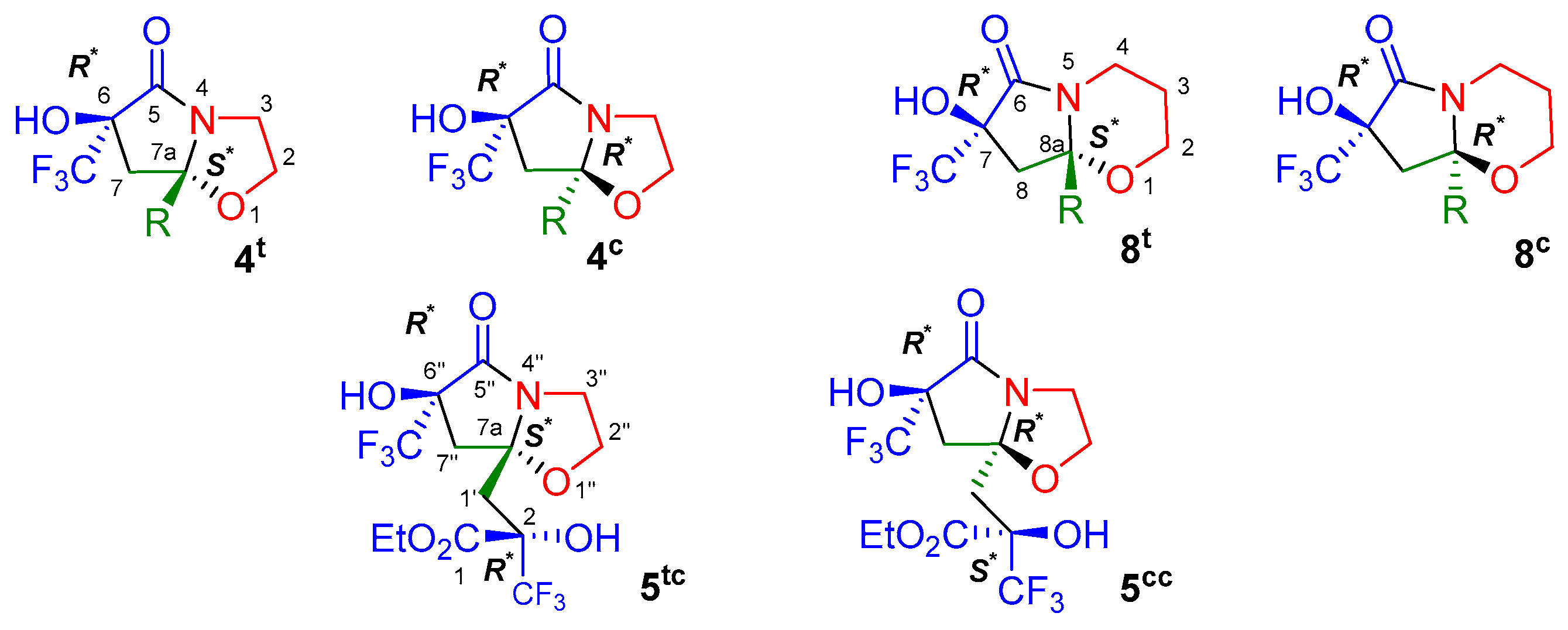
| Entry | Conditions * | 1 (eq) | Time, Day | T, °C | Composition of the Reaction Mixture According to 19F NMR Data, δ, ppm ** | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 4ac | 4at | 5tc | 5cc | 6a | 7a | By-Prod. | |||||
| 1 | 1,4-dioxane | 1 | 7 | 25 | 26 | 40 | 11 | 12 | - | - | 11 |
| 2 | Toluene | 1 | 7 | 25 | 26 | 27 | 17 | 18 | - | - | 12 |
| 3 | EtOH | 1 | 7 | 25 | 23 | 26 | 12 | 16 | - | - | 23 |
| 4 | C2H4Cl2 | 1 | 7 | 25 | 23 | 25 | 16 | 15 | - | - | 21 |
| 5 | MeCN | 1 | 7 | 25 | 15 | 30 | 21 | 20 | - | - | 12 |
| 6 | THF | 1 | 7 | 25 | 13 | 29 | 20 | 22 | - | - | 16 |
| 7 | C2H4Cl2 | 2 | 7 | 25 | 6 | 5 | 32 | 30 | 8 | 4 | 18 |
| 8 | 1,4-dioxane | 2 | 7 | 25 | 3 | 4 | 35 | 37 | 3 | 4 | 14 |
| 9 | THF | 2 | 7 | 25 | 2 | 3 | 37 | 40 | 2 | 10 | 6 |
| 10 | THF | 2 | 2 | 50 | 6 | 4 | 28 | 31 | - | - | 31 |
| Products (δF, ppm), 19F NMR Data | The Ratio of Diastereomers in the Reaction Mixture, % | |
|---|---|---|
| Three-Component Method | Two-Component Method | |
| 4ac (83.63):4at (83.91) | 39:61 | 16:84 |
| 4bc (83.79):4bt (83.89) | 44:56 | 22:78 |
| 8ac (83.71):8at (84.22) | 56:44 | 65:35 |
| 8bc (83.85):8bt (84.14) | 54:46 | 67:33 |
| Compounds | R | δH-A, ppm | δH-B, ppm | ΔAB, ppm | 2J, Hz |
|---|---|---|---|---|---|
| 4ac | Me | 2.74 | 2.29 | 0.45 | 15.2 |
| 4at | Me | 2.37 | 2.33 | 0.04 | 14.2 |
| 4bc | Et | 2.73 | 2.17 | 0.56 | 15.5 |
| 4bt | Et | 2.42 | 2.23 | 0.19 | 14.5 |
| 4cc | Bu | 2.73 | 2.19 | 0.54 | 15.4 |
| 4ct | Bu | 2.42 | 2.24 | 0.18 | 14.5 |
| 4dc | Ph | 2.80 | 2.70 | 0.10 | 15.5 |
| 8ac | Me | 2.56 | 2.15 | 0.41 | 15.2 |
| 8at | Me | 2.30 | 2.23 | 0.07 | 14.4 |
| 8bc | Et | 2.52 | 2.00 | 0.52 | 15.3 |
| 8bt | Et | 2.33 | 2.10 | 0.23 | 14.8 |
| 8cc | Bu | 2.52 | 2.02 | 0.50 | 15.3 |
| 8ct | Bu | 2.33 | 2.12 | 0.21 | 14.7 |
| 8dt | Ph | 2.64 | 2.22 | 0.42 | 14.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goryaeva, M.V.; Fefelova, O.A.; Burgart, Y.V.; Ezhikova, M.A.; Kodess, M.I.; Slepukhin, P.A.; Gaviko, V.S.; Saloutin, V.I. Multicomponent Domino Cyclization of Ethyl Trifluoropyruvate with Methyl Ketones and Amino Alcohols as A New Way to γ-Lactam Annulated Oxazacycles. Molecules 2023, 28, 1983. https://doi.org/10.3390/molecules28041983
Goryaeva MV, Fefelova OA, Burgart YV, Ezhikova MA, Kodess MI, Slepukhin PA, Gaviko VS, Saloutin VI. Multicomponent Domino Cyclization of Ethyl Trifluoropyruvate with Methyl Ketones and Amino Alcohols as A New Way to γ-Lactam Annulated Oxazacycles. Molecules. 2023; 28(4):1983. https://doi.org/10.3390/molecules28041983
Chicago/Turabian StyleGoryaeva, Marina V., Olesya A. Fefelova, Yanina V. Burgart, Marina A. Ezhikova, Mikhail I. Kodess, Pavel A. Slepukhin, Vasily S. Gaviko, and Victor I. Saloutin. 2023. "Multicomponent Domino Cyclization of Ethyl Trifluoropyruvate with Methyl Ketones and Amino Alcohols as A New Way to γ-Lactam Annulated Oxazacycles" Molecules 28, no. 4: 1983. https://doi.org/10.3390/molecules28041983
APA StyleGoryaeva, M. V., Fefelova, O. A., Burgart, Y. V., Ezhikova, M. A., Kodess, M. I., Slepukhin, P. A., Gaviko, V. S., & Saloutin, V. I. (2023). Multicomponent Domino Cyclization of Ethyl Trifluoropyruvate with Methyl Ketones and Amino Alcohols as A New Way to γ-Lactam Annulated Oxazacycles. Molecules, 28(4), 1983. https://doi.org/10.3390/molecules28041983