
Phosphorus-Containing Polymers as Sensitive Biocompatible Probes for 31P Magnetic Resonance

 , and
, and
Abstract
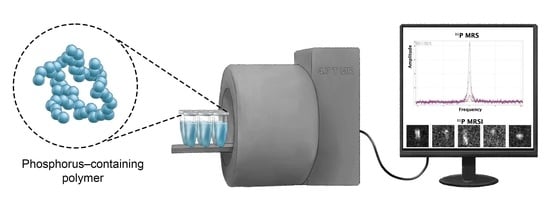
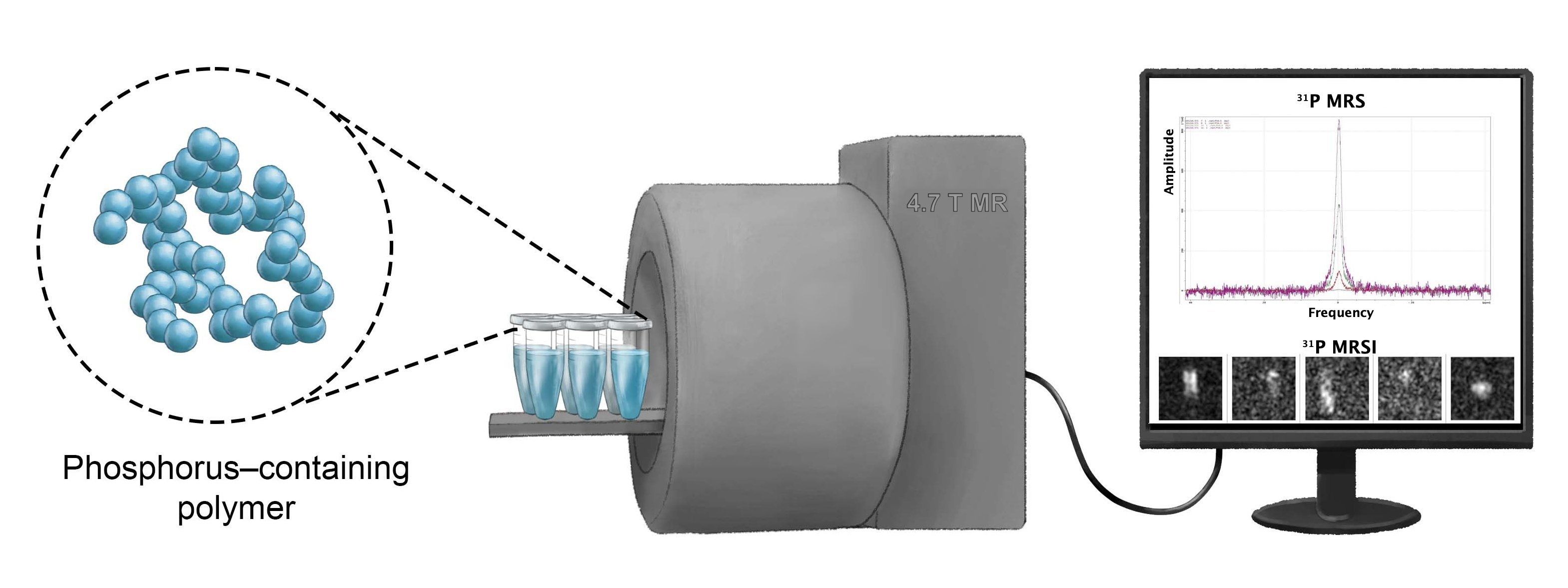
:
1. Introduction
2. Results and Discussion
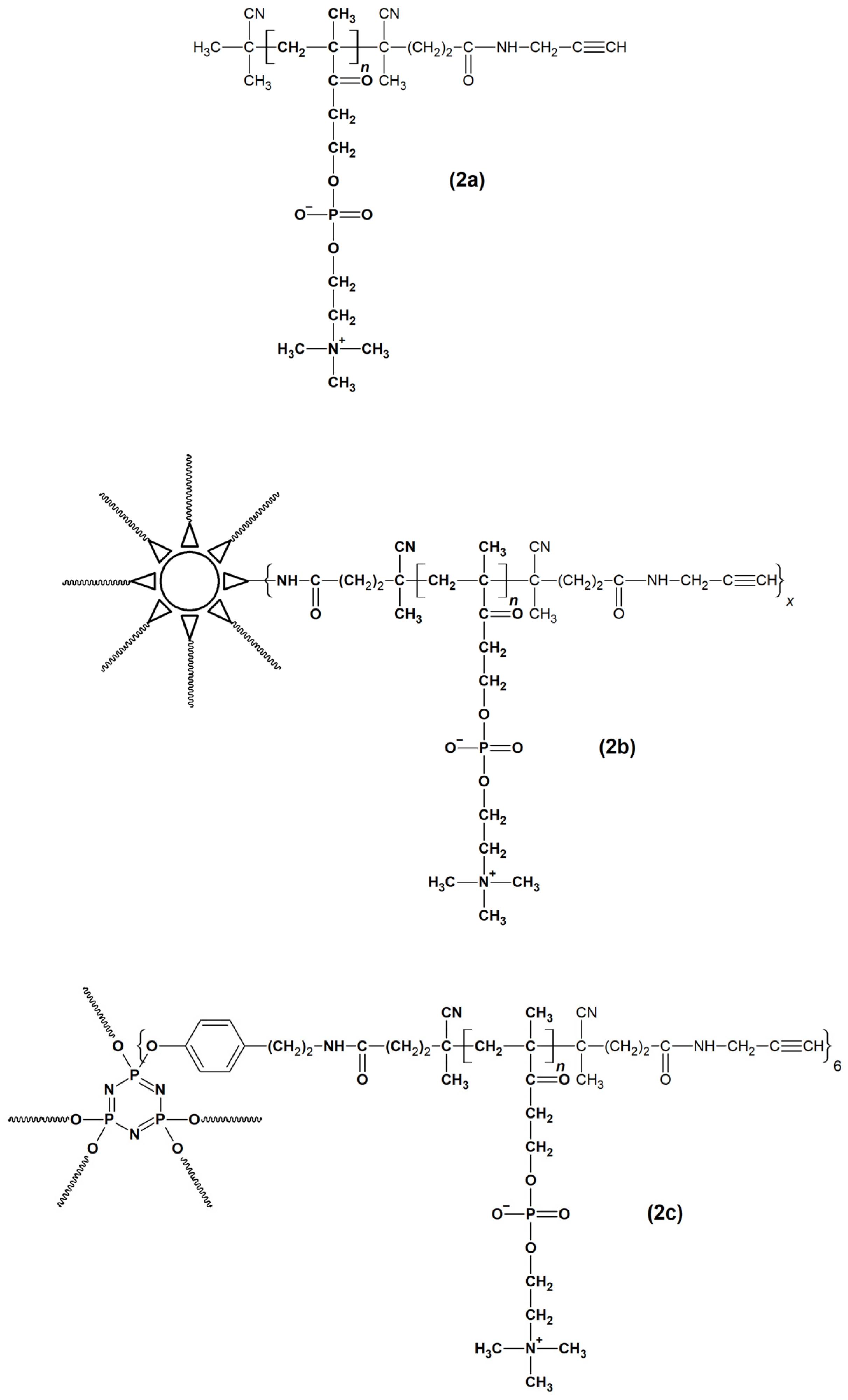
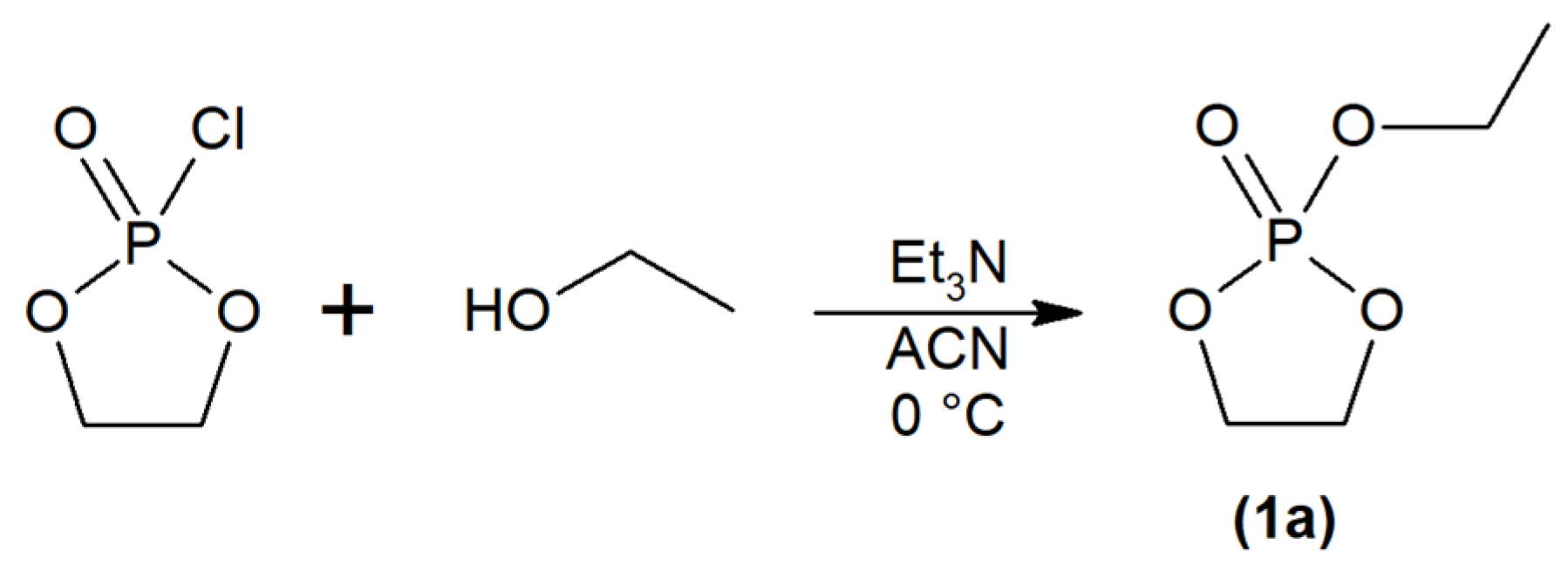
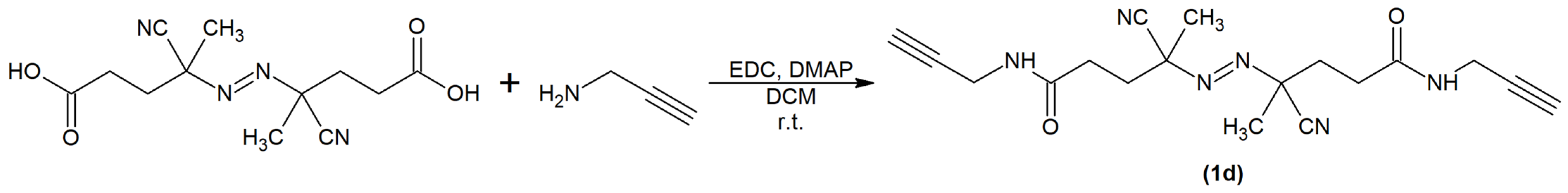
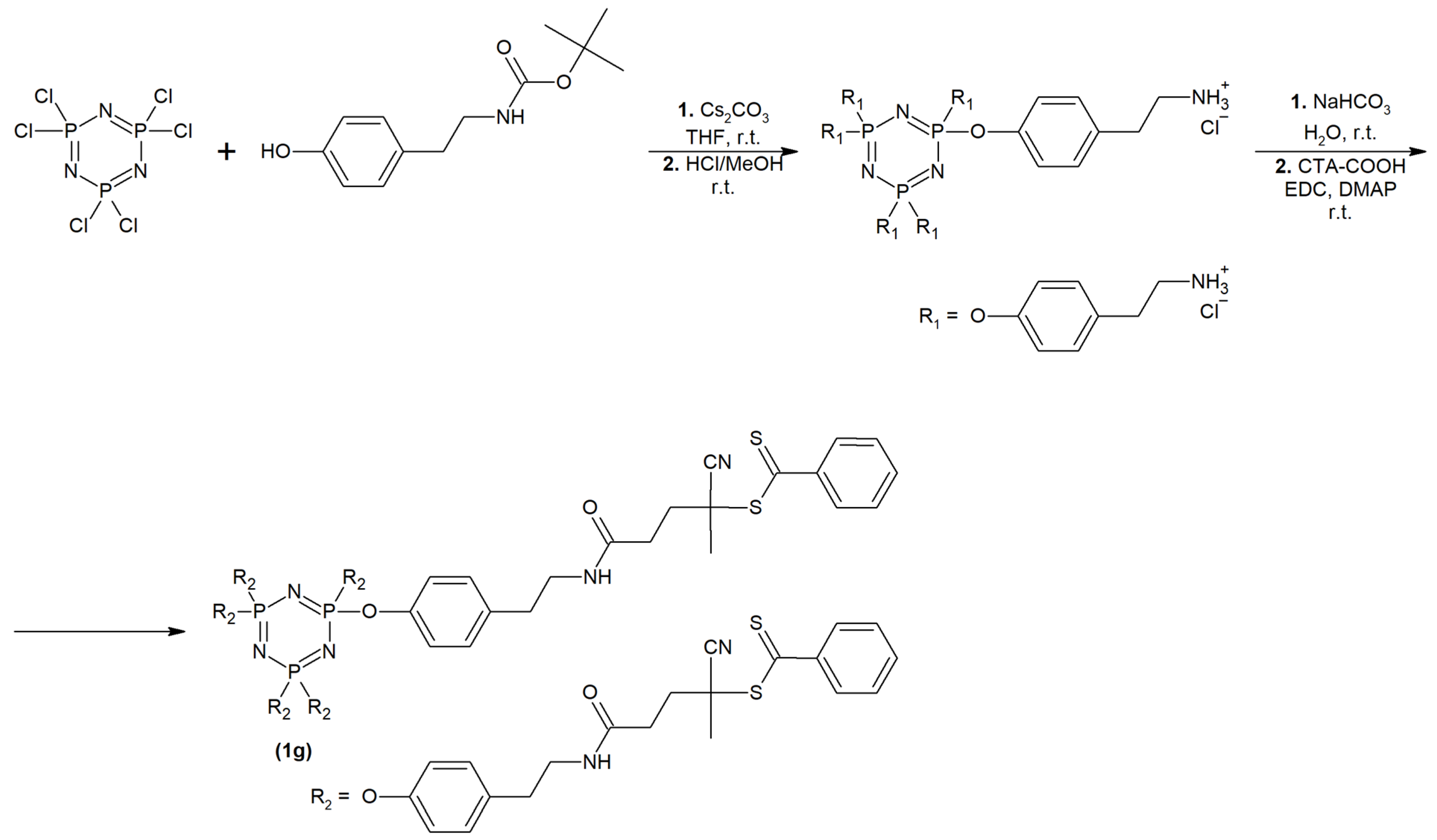
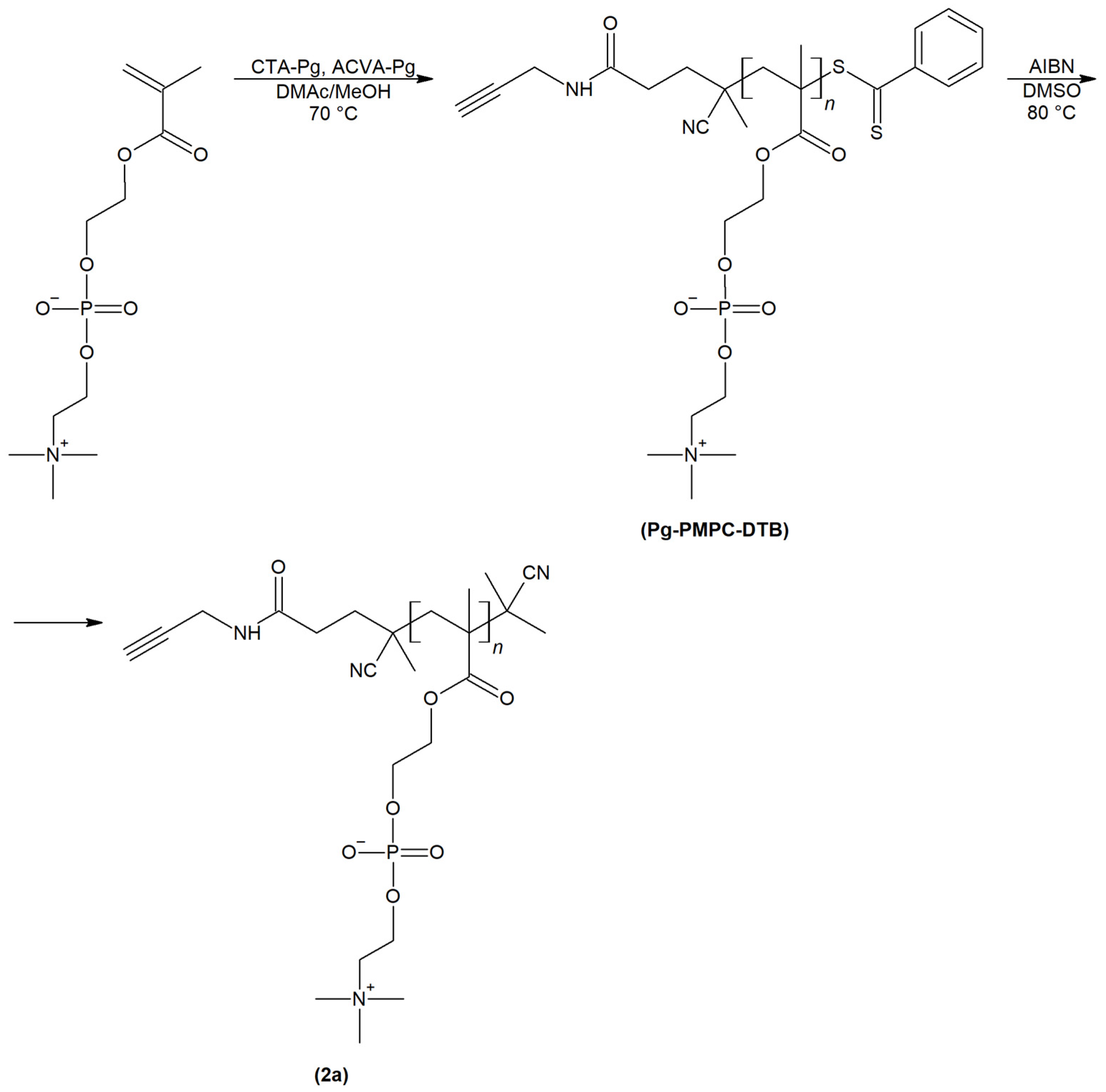
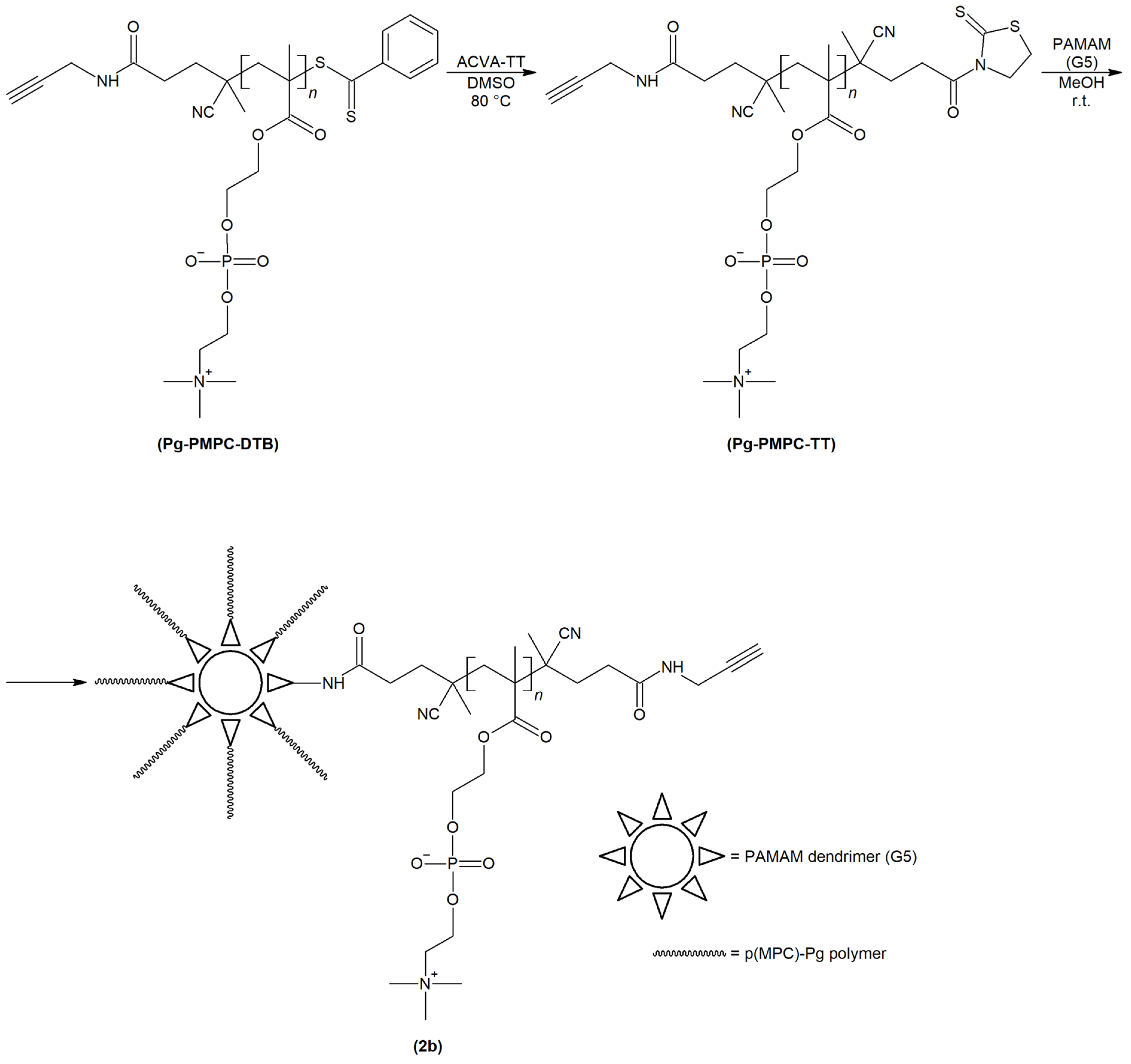
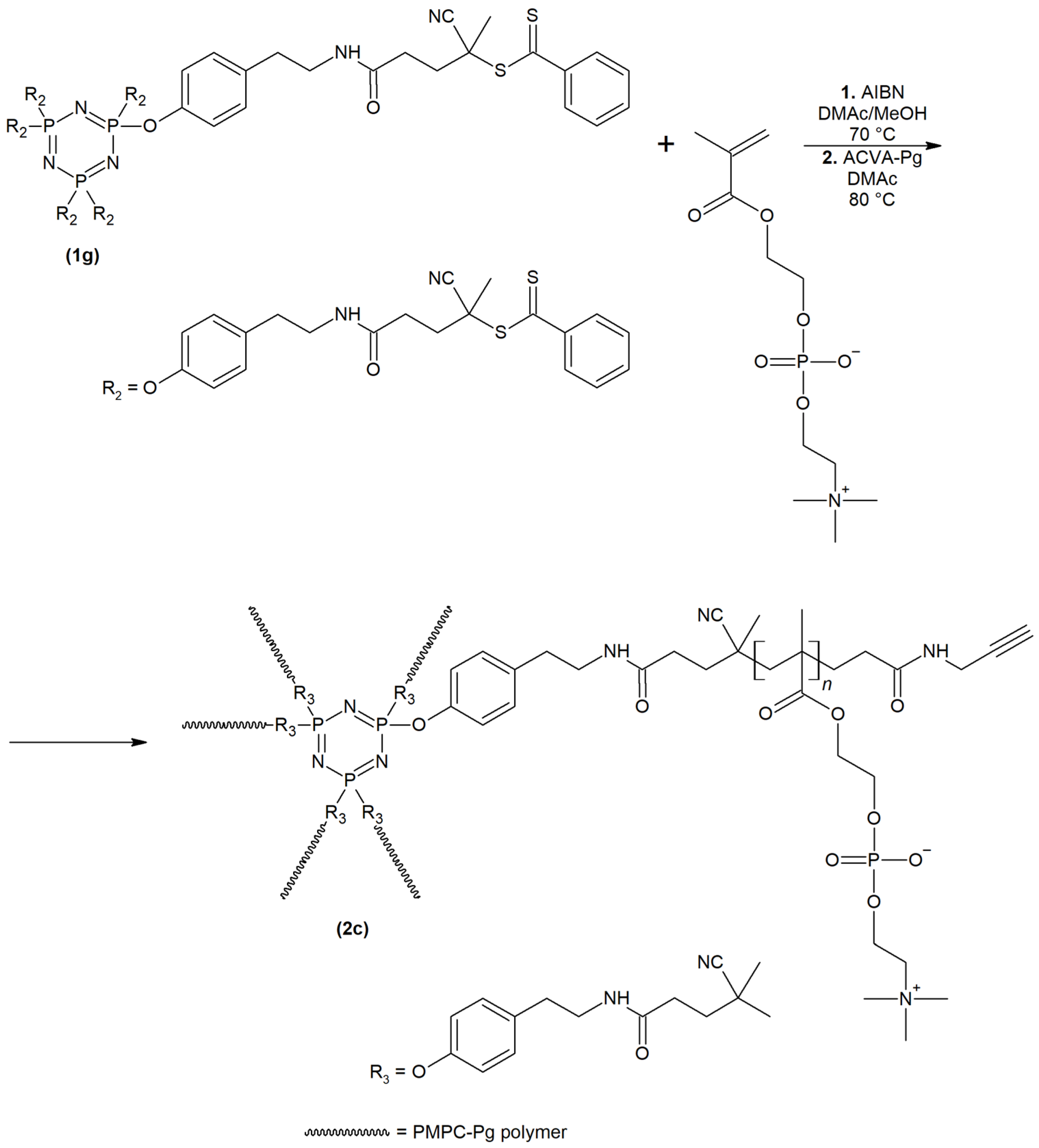
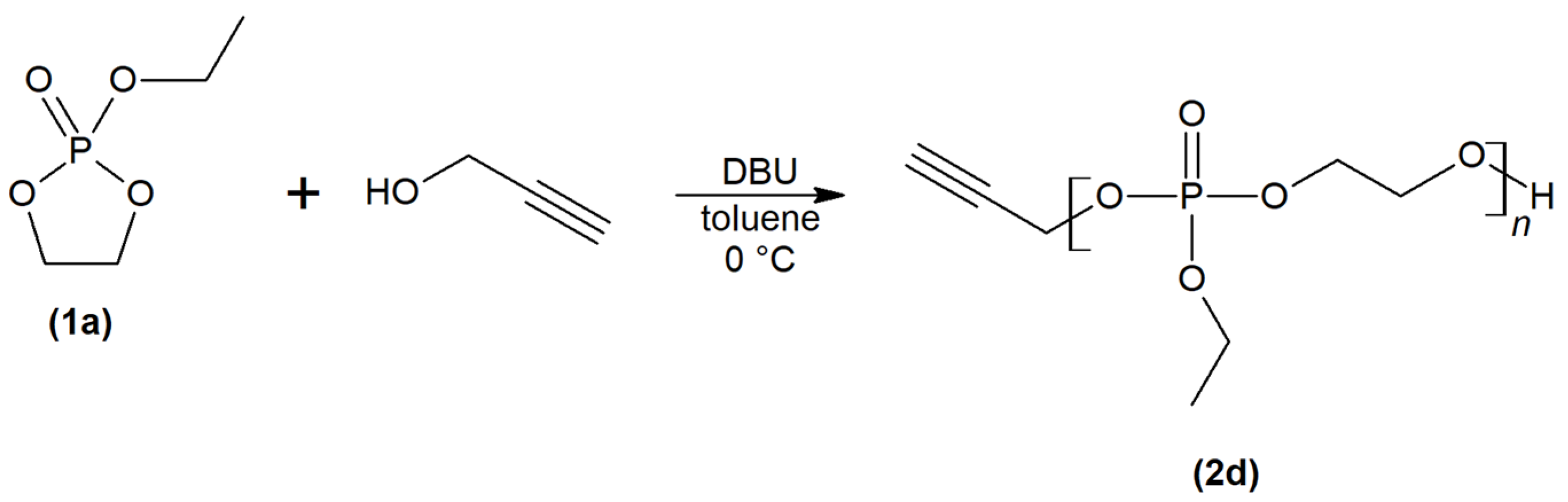
2.1. Synthesis and Physiochemical Characterization of Polymer Probes
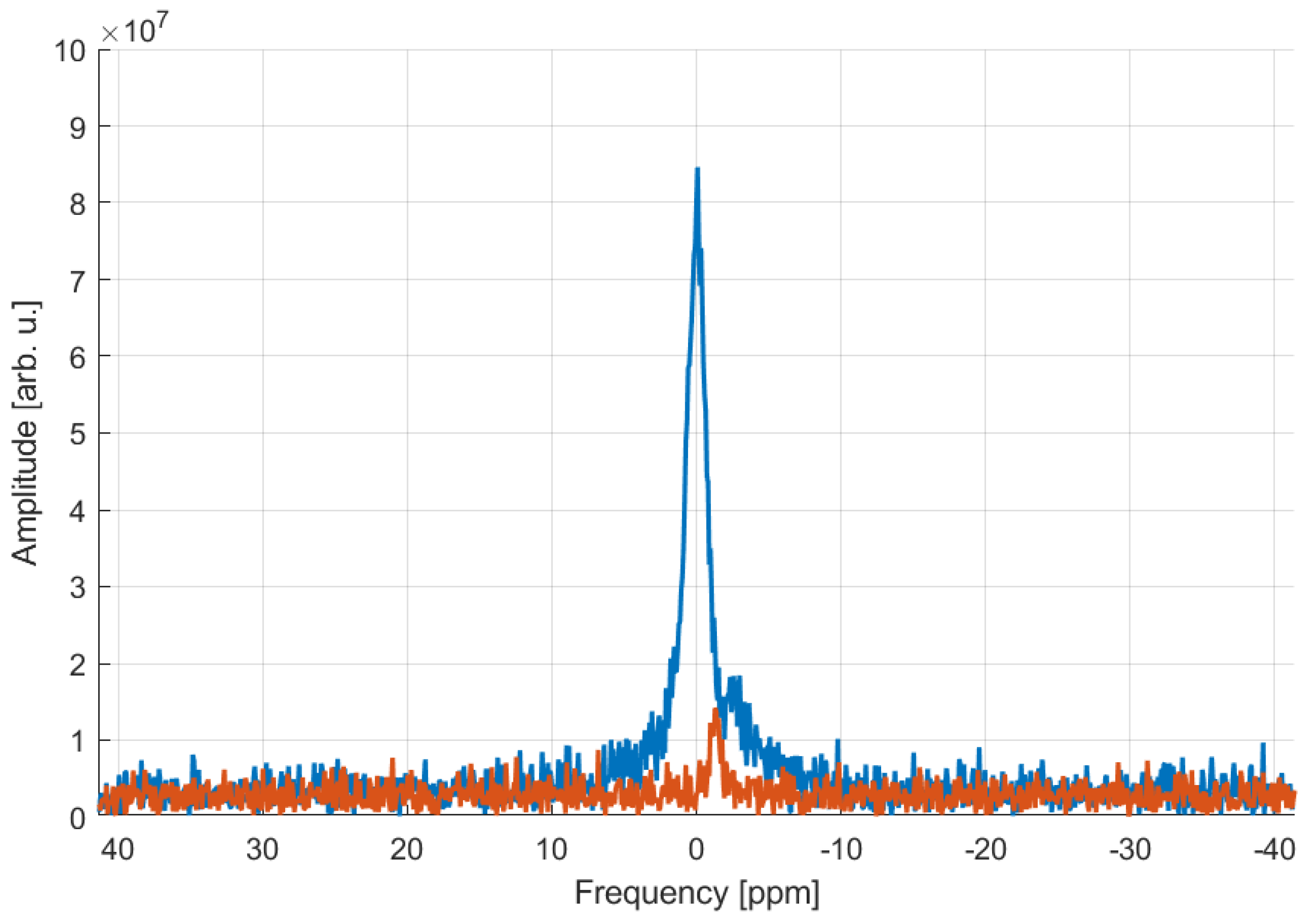
2.2. MR Properties of Polymer Probes
3. Materials
3.1. Chemicals
3.2. Synthesis of Monomers, Initiators and Chain Transfer Agents
3.3. Synthesis of Polymer Probes
4. Methods
4.1. UV–Vis Spectrophotometry
4.2. Size-exclusion Chromatography
4.3. High-performance Liquid Chromatography
4.4. Nuclear Magnetic Resonance
4.5. Magnetic Resonance Spectroscopy, Imaging and Relaxometry
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Santos-Díaz, A.; Noseworthy, M.D. Phosphorus magnetic resonance spectroscopy and imaging (31P-MRS/MRSI) as a window to brain and muscle metabolism: A review of the methods. Biomed. Signal. Process. Control. 2020, 60, 101967. [Google Scholar] [CrossRef]
- Ruhm, L.; Dorst, J.; Avdievitch, N.; Wright, A.M.; Henning, A. 3D 31P MRSI of the human brain at 9.4 Tesla: Optimization and quantitative analysis of metabolic images. Magn. Reson. Med. 2021, 86, 2368–2383. [Google Scholar]
- Wijnen, J.P.; Scheenen, T.W.J.; Klomp, D.W.J.; Heerschap, A. 31P Magnetic resonance spectroscopic imaging with polarisation transfer of phosphomono- and diesters at 3 T in the human brain: Relation with age and spatial differences. NMR Biomed. 2010, 23, 968–976. [Google Scholar]
- Liu, Y.; Gu, Y.; Yu, X. Assessing tissue metabolism by phosphorous-31 magnetic resonance spectroscopy and imaging: A methodology review. Quant. Imaging Med. Surg. 2017, 7, 707–726. [Google Scholar]
- Neeman, M.; Rushkin, E.; Kaye, A.M.; Degani, H. 31P-NMR studies of phosphate transfer rates in T47D human breast cancer cells. Biochimica Biophysica Acta (BBA)-Mol. Cell Res. 1987, 930, 179–192. [Google Scholar]
- Scheuermann-Freestone, M.; Madsen, P.L.; Manners, D.; Blamire, A.M.; Buckingham, R.E.; Styles, P.; Radda, G.K.; Neubauer, S.; Clarke, K. Abnormal Cardiac and Skeletal Muscle Energy Metabolism in Patients With Type 2 Diabetes. Circulation 2003, 107, 3040–3046. [Google Scholar]
- Levine, S.R.; Helpern, J.A.; Welch, K.M.; Linde, A.M.V.; Sawaya, K.L.; Brown, E.E.; Ramadan, N.M.; Deveshwar, R.K.; Ordidge, R.J. Human focal cerebral ischemia: Evaluation of brain pH and energy metabolism with P-31 NMR spectroscopy. Radiology 1992, 185, 537–544. [Google Scholar]
- Weiss, R.G.; Gerstenblith, G.; Bottomley, P.A. ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proc. Natl. Acad. Sci. USA 2005, 102, 808–813. [Google Scholar]
- Kemp, G.J.; Meyerspeer, M.; Moser, E. Absolute quantification of phosphorus metabolite concentrations in human muscle in vivo by 31P MRS: A quantitative review. NMR Biomed. 2007, 20, 555–565. [Google Scholar]
- Zhao, K.; Li, D.; Shi, C.; Ma, X.; Rong, G.; Kang, H.; Wang, X.; Sun, B. Biodegradable Polymeric Nanoparticles as the Delivery Carrier for Drug. Curr. Drug Deliv. 2016, 13, 494–499. [Google Scholar]
- Prajapati, S.K.; Jain, A.; Jain, A.; Jain, S. Biodegradable polymers and constructs: A novel approach in drug delivery. Eur. Polym. J. 2019, 120, 109191. [Google Scholar] [CrossRef]
- Ogueri, K.S.; Ogueri, K.S.; Ude, C.C.; Allcock, H.R.; Laurencin, C.T. Biomedical applications of polyphosphazenes. Med. Devices Sens. 2020, 3, e10113. [Google Scholar]
- Andrianov, A.K. Water-Soluble Polyphosphazenes for Biomedical Applications. J. Inorg. Organomet. Polym. Mater. 2006, 16, 397–406. [Google Scholar]
- Zhang, S.; Ali, S.; Ma, H.; Zhang, L.; Wu, Z.; Wu, D.; Hu, T.S. Preparation of Poly(bis(phenoxy)phosphazene) and 31P NMR Analysis of Its Structural Defects under Various Synthesis Conditions. J. Phys. Chem. B 2016, 120, 11307–11316. [Google Scholar]
- Weikel, A.L.; Owens, S.G.; Fushimi, T.; Allcock, H.R. Synthesis and Characterization of Methionine- and Cysteine-Substituted Phosphazenes. Macromolecules 2010, 43, 5205–5210. [Google Scholar]
- Yilmaz, Z.E.; Jérôme, C. Polyphosphoesters: New Trends in Synthesis and Drug Delivery Applications. Macromol. Biosci. 2016, 16, 1745–1761. [Google Scholar]
- Pelosi, C.; Tinè, M.R.; Wurm, F.R. Main-chain water-soluble polyphosphoesters: Multi-functional polymers as degradable PEG-alternatives for biomedical applications. Eur. Polym. J. 2020, 141, 110079. [Google Scholar] [CrossRef]
- Goda, T.; Ishihara, K.; Miyahara, Y. Critical update on 2-methacryloyloxyethyl phosphorylcholine (MPC) polymer science. J. Appl. Polym. Sci. 2015, 132, 132. [Google Scholar] [CrossRef]
- Kojima, C.; Katayama, R.; Lien Nguyen, T.; Oki, Y.; Tsujimoto, A.; Yusa, S.-I.; Shiraishi, K.; Matsumoto, A. Different antifouling effects of random and block copolymers comprising 2-methacryloyloxyethyl phosphorylcholine and dodecyl methacrylate. Eur. Polym. J. 2020, 136, 109932. [Google Scholar] [CrossRef]
- Nazarova, O.; Chernova, E.; Dobrodumov, A.; Zolotova, Y.; Bezrukova, M.; Nekrasova, T.; Vlasova, E.; Panarin, E. New water-soluble copolymers of 2-methacryloyloxyethyl phosphorylcholine for surface modification. J. Appl. Polym. Sci. 2021, 138, 50272. [Google Scholar] [CrossRef]
- Chen, Y.; Diaz-Dussan, D.; Wu, D.; Wang, W.; Peng, Y.-Y.; Asha, A.B.; Hall, D.G.; Ishihara, K.; Narain, R. Bioinspired Self-Healing Hydrogel Based on Benzoxaborole-Catechol Dynamic Covalent Chemistry for 3D Cell Encapsulation. ACS Macro Lett. 2018, 7, 904–908. [Google Scholar]
- Kracíková, L.; Ziółkowska, N.; Androvič, L.; Klimánková, I.; Červený, D.; Vít, M.; Pompach, P.; Konefał, R.; Janoušková, O.; Hrubý, M.; et al. Phosphorus-Containing Polymeric Zwitterion: A Pioneering Bioresponsive Probe for 31P-Magnetic Resonance Imaging. Macromol. Biosci. 2022, 22, 2100523. [Google Scholar] [CrossRef]
- Oatway, L.; Vasanthan, T.; Helm, J.H. Phytic Acid. Food Rev. Int. 2001, 17, 419–431. [Google Scholar]
- Ziółkowska, N.; Vít, M.; Laga, R.; Jirák, D. Iron-doped calcium phytate nanoparticles as a bio-responsive contrast agent in 1H/31P magnetic resonance imaging. Sci. Rep. 2022, 12, 2118. [Google Scholar] [CrossRef]
- Francescato, M.P.; Cettolo, V.; di Prampero, P.E. Influence of phosphagen concentration on phosphocreatine breakdown kinetics. Data from human gastrocnemius muscle. J. Appl. Physiol. 2008, 105, 158–164. [Google Scholar]
- Clément, B.; Grignard, B.; Koole, L.; Jérôme, C.; Lecomte, P. Metal-Free Strategies for the Synthesis of Functional and Well-Defined Polyphosphoesters. Macromolecules 2012, 45, 4476–4486. [Google Scholar]
- Wang, B.; Rivard, E.; Manners, I. A new high-yield synthesis of Cl(3)P=NSiMe(3), a monomeric precursor for the controlled preparation of high molecular weight polyphosphazenes. Inorg. Chem. 2002, 41, 1690–1691. [Google Scholar]
- Wilfert, S.; Henke, H.; Schöfberger, W.; Brüggemann, O.; Teasdale, I. Chain-End-Functionalized Polyphosphazenes via a One-Pot Phosphine-Mediated Living Polymerization. Macromol. Rapid Commun. 2014, 35, 1135–1141. [Google Scholar]
- Šubr, V.; Kostka, L.; Strohalm, J.; Etrych, T.; Ulbrich, K. Synthesis of Well-Defined Semitelechelic Poly[N-(2-hydroxypropyl)methacrylamide] Polymers with Functional Group at the α-End of the Polymer Chain by RAFT Polymerization. Macromolecules 2013, 46, 2100–2108. [Google Scholar]
- Šubr, V.; Konák, C.; Laga, R.; Ulbrich, K. Coating of DNA/Poly(l -lysine) Complexes by Covalent Attachment of Poly[ N -(2-hydroxypropyl)methacrylamide]. Biomacromolecules 2006, 7, 122–130. [Google Scholar]
- Androvič, L.; Woldřichová, L.; Jozefjaková, K.; Pechar, M.; Lynn, G.M.; Kaňková, D.; Malinová, L.; Laga, R. Cyclotriphosphazene-Based Star Copolymers as Structurally Tunable Nanocarriers with Programmable Biodegradability. Macromolecules 2021, 54, 3139–3157. [Google Scholar]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Polymer Probe | (2a) | (2b) | (2c) | (2d) | (2e) |
|---|---|---|---|---|---|
| Structure | IBN-PMPC-Pg | PAMAM-g-PMPC-Pg | CTP-g-PMPC-Pg | PEEP-Pg | PMEEEP-Vi |
| Mn [kg·mol−1] | 18.8 | 398.8 | 58.6 | 3.7 | 22.0 |
| Ð | 1.06 | 1.10 | 1.10 | 1.03 | 1.20 |
| SNR (31P MRSI) | 3.52 | 1.39 | 1.54 | 1.11 | 10.4 |
| SNR (31P MRS) | 209.8 | 55.7 | 43.6 | 12.1 | 61.6 |
| 31P T1 [ms] | 1078.2 | 1433.7 | 1454.1 | 2367.8 | 1139.4 |
| 31P T2 [ms] | 58.3 | 78.3 | 170.6 | 113.4 | 30.3 |
| 31P MRSI |  |  |  |  |  |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kracíková, L.; Androvič, L.; Potočková, I.; Ziółkowska, N.; Vít, M.; Červený, D.; Jirák, D.; Laga, R. Phosphorus-Containing Polymers as Sensitive Biocompatible Probes for 31P Magnetic Resonance. Molecules 2023, 28, 2334. https://doi.org/10.3390/molecules28052334
Kracíková L, Androvič L, Potočková I, Ziółkowska N, Vít M, Červený D, Jirák D, Laga R. Phosphorus-Containing Polymers as Sensitive Biocompatible Probes for 31P Magnetic Resonance. Molecules. 2023; 28(5):2334. https://doi.org/10.3390/molecules28052334
Chicago/Turabian StyleKracíková, Lucie, Ladislav Androvič, Iveta Potočková, Natalia Ziółkowska, Martin Vít, David Červený, Daniel Jirák, and Richard Laga. 2023. "Phosphorus-Containing Polymers as Sensitive Biocompatible Probes for 31P Magnetic Resonance" Molecules 28, no. 5: 2334. https://doi.org/10.3390/molecules28052334
APA StyleKracíková, L., Androvič, L., Potočková, I., Ziółkowska, N., Vít, M., Červený, D., Jirák, D., & Laga, R. (2023). Phosphorus-Containing Polymers as Sensitive Biocompatible Probes for 31P Magnetic Resonance. Molecules, 28(5), 2334. https://doi.org/10.3390/molecules28052334