
Diphenyl Diselenide-Assisted Radical Addition Reaction of Diphenyl Disulfide to Unsaturated Bonds upon Photoirradiation


Abstract
:1. Introduction
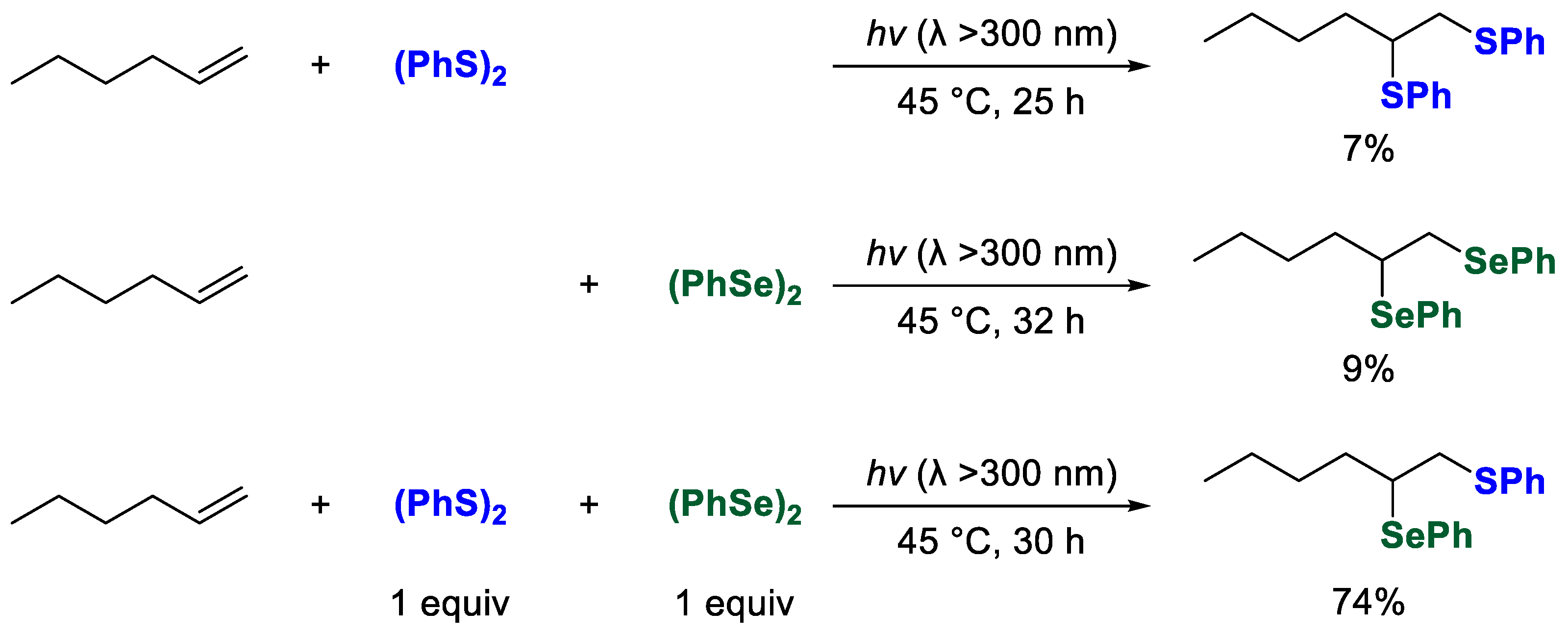
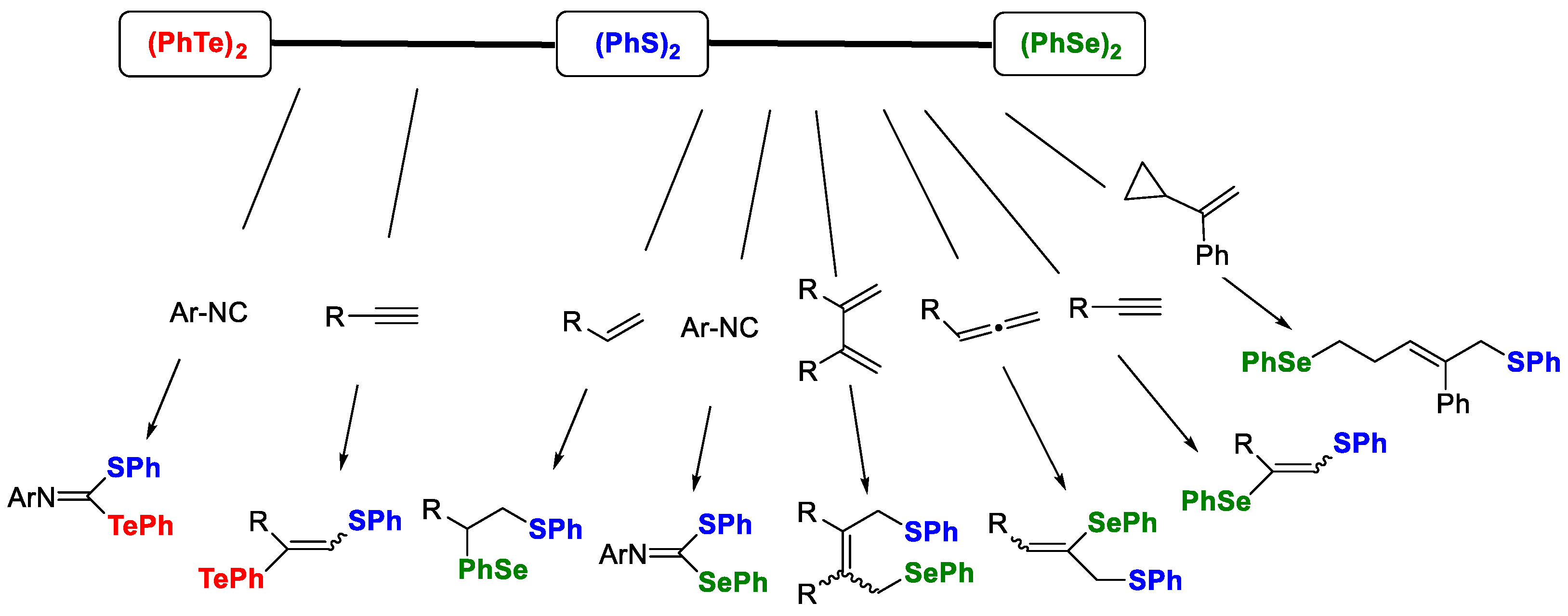
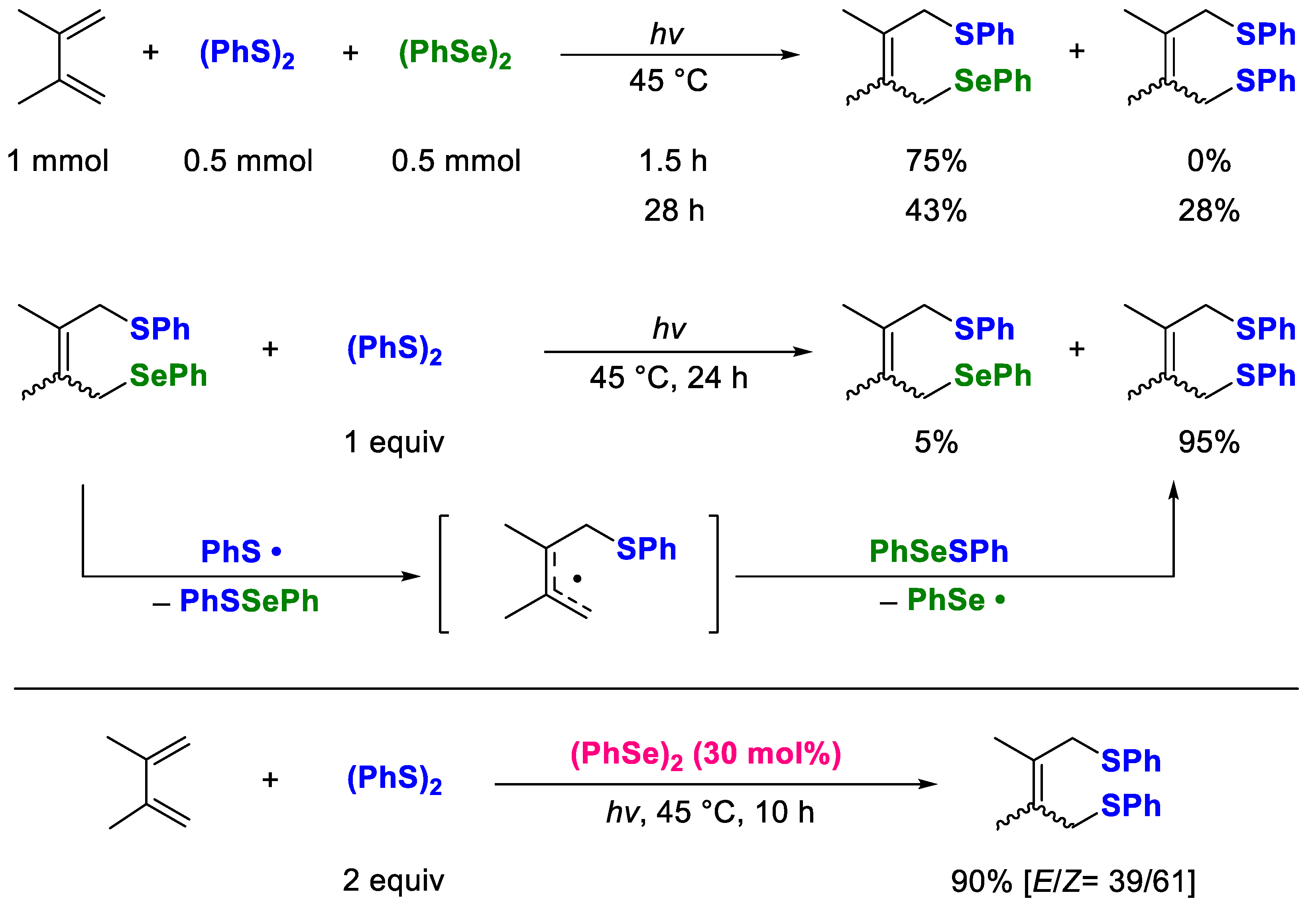
2. Results and Discussion
 | (1) |
 | (2) |
 | (3) |
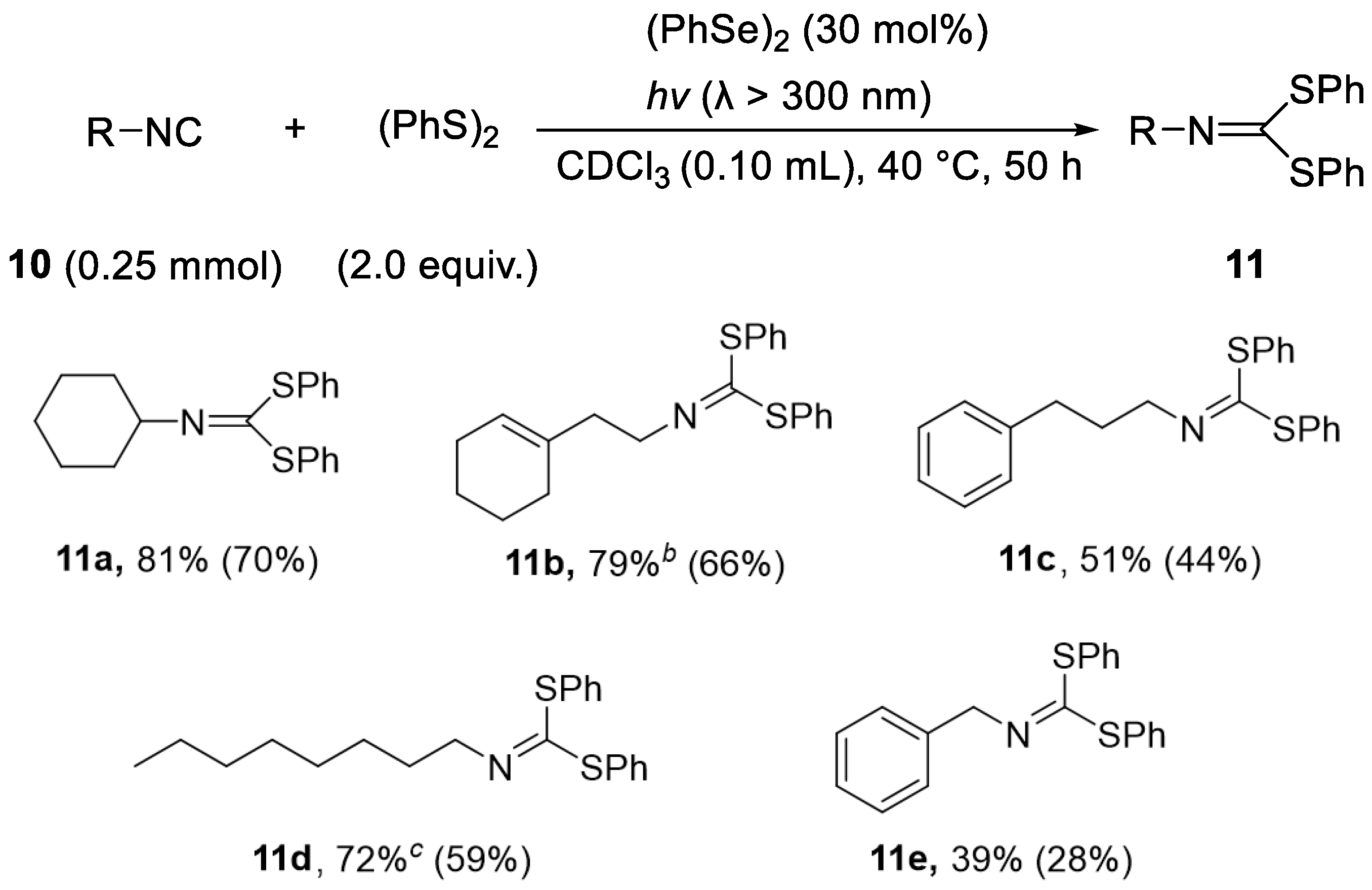
 | (4) |
 | (5) |
3. Materials and Methods
3.1. General Information
3.2. (PhSe)2-Assisted Dithiolation of 2,3-Dihydrofuran 1k under Photoirradiation
3.3. (PhSe)2-Assisted Dithiolation of Styrene 1j under Photoirradiation
3.4. (PhTe)2-Assisted Dithiolation of Norbornene 1h under Photoirradiation
3.5. General Procedure for the (PhSe)2-Catalyzed Dithiolation of Isocyanides
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Studer, A.; Curran, D.P. Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem. Int. Ed. 2016, 55, 58–102. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Xu, L.; Porter, N.A. Free Radical Lipid Peroxidation: Mechanisms and Analysis. Chem. Rev. 2011, 111, 5944–5972. [Google Scholar] [CrossRef] [PubMed]
- Dénès, F.; Pichowicz, M.; Povie, G.; Renaud, P. Thiyl Radicals in Organic Synthesis. Chem. Rev. 2014, 114, 2587–2693. [Google Scholar] [CrossRef] [PubMed]
- Ollivier, C.; Renaud, P. Organoboranes as a Source of Radicals. Chem. Rev. 2001, 101, 3415–3434. [Google Scholar] [CrossRef]
- Kamigaito, M.; Ando, T.; Sawamoto, M. Metal-Catalyzed Living Radical Polymerization. Chem. Rev. 2001, 101, 3689–3745. [Google Scholar] [CrossRef]
- Pearson, R.M.; Lim, C.H.; McCarthy, B.G.; Musgrave, C.B.; Miyake, G.M. Organocatalyzed Atom Transfer Radical Polymerization Using N-Aryl Phenoxazines as Photoredox Catalysts. J. Am. Chem. Soc. 2016, 138, 11399–11407. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.Y.; Zhao, Q.Q.; Chen, J.; Xiao, W.J.; Chen, J.R. When Light Meets Nitrogen-Centered Radicals: From Reagents to Catalysts. Acc. Chem. Res. 2020, 53, 1066–1083. [Google Scholar] [CrossRef]
- McCarthy, B.G.; Pearson, R.M.; Lim, C.H.; Sartor, S.M.; Damrauer, N.H.; Miyake, G.M. Structure-Property Relationships for Tailoring Phenoxazines as Reducing Photoredox Catalysts. J. Am. Chem. Soc. 2018, 140, 5088–5510. [Google Scholar] [CrossRef]
- Maeda, H.; Maeda, T.; Mizuno, K.; Fujimoto, K.; Shimizu, H.; Inouye, M. Alkynylpyrenes as Improved Pyrene-Based Biomolecular Probes with the Advantages of High Fluorescence Quantum Yields and Long Absorption/Emission Wavelengths. Chem. Eur. J. 2006, 12, 824–831. [Google Scholar] [CrossRef]
- Ryu, I.; Kreimerman, S.; Araki, F.; Nishitani, S.; Oderaotoshi, Y.; Minakata, S.; Komatsu, M. Cascade Radical Reactions Catalyzed by a Pd/Light System: Cyclizative Multiple Carbonylation of 4-Alkenyl Iodides. J. Am. Chem. Soc. 2002, 124, 3812–3813. [Google Scholar] [CrossRef]
- Ogawa, A. 4.06 Addition of X–Y Reagents to Alkenes, Alkynes, and Allenes. Compr. Org. Synth. Second Ed. 2014, 4, 392–411. [Google Scholar]
- Yamamoto, Y.; Ogawa, A. Metal-Free One-Pot Multi-Functionalization of Unsaturated Compounds with Interelement Compounds by Radical Process. Molecules 2023, 28, 787. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Koguma, Y.; Tanaka, T.; Umeda, R. Cesium Carbonate-Catalyzed α-Phenylchalcogenation of Carbonyl Compounds with Diphenyl Dichalcogenide. Molecules 2009, 14, 3367–3375. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Garcia, J.M.; Haeffner, F.; Radomkit, S.; Zhugralin, A.R.; Hoveyda, A.H. Mechanism of NHC-Catalyzed Conjugate Additions of Diboron and Borosilane Reagents to α,β-Unsaturated Carbonyl Compounds. J. Am. Chem. Soc. 2015, 137, 10585–10602. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Montgomery, J. Functionalization of Styrenes by Copper-Catalyzed Borylation/ortho-Cyanation and Silver-Catalyzed Annulation Processes. Angew. Chem. Int. Ed. 2015, 54, 12683–12686. [Google Scholar] [CrossRef]
- Zhao, B.; Li, Z.; Wu, Y.; Wang, Y.; Qian, J.; Yuan, Y.; Shi, Z. An Olefinic 1,2-Boryl-Migration Enabled by Radical Addition: Construction of gem-Bis(Boryl)alkanes. Angew. Chem. Int. Ed. 2019, 58, 9448–9452. [Google Scholar] [CrossRef]
- Wang, D.; Mück-Lichtenfeld, C.; Studer, A. 1,n-Bisborylalkanes via Radical Boron Migration. J. Am. Chem. Soc. 2020, 142, 9119–9123. [Google Scholar] [CrossRef]
- Taniguchi, T.; Fujii, T.; Idota, A.; Ishibashi, H. Reductive Addition of the Benzenethiyl Radical to Alkynes by Amine-Mediated Single Electron Transfer Reaction to Diphenyl Disulfide. Org. Lett. 2009, 11, 3298–3301. [Google Scholar] [CrossRef]
- Heiba, E.I.; Dessau, R.M. Free-Radical Addition of Organic Disulfides to Acetylenes. J. Org. Chem. 1967, 32, 3837–3840. [Google Scholar] [CrossRef]
- Borghi, R.; Lunazzi, L.; Placucci, G.; Cerioni, G.; Plumitallo, A. Photolysis of Dialkoxy Disulfides: A Convenient Source of Alkoxy Radicals for Addition to the Sphere of Fullerene C60. J. Org. Chem. 1996, 61, 3327–3331. [Google Scholar] [CrossRef]
- Back, T.G.; Krishna, M.V. Free-Radical Additions of Diselenides to Dimethyl Acetylenedicarboxylate, Methyl Propiolate, and Dimethyl Maleate. J. Org. Chem. 1988, 53, 2533–2536. [Google Scholar] [CrossRef]
- Kwak, Y.; Tezuka, M.; Goto, A.; Fukuda, T.; Yamago, S. Kinetic Study on Role of Ditelluride in Organotellurium-Mediated Living Radical Polymerization (TERP). Macromolecules 2007, 40, 1881–1885. [Google Scholar] [CrossRef]
- Ogawa, A.; Yokoyama, K.; Yokoyama, H.; Obayashi, R.; Kambe, N.; Sonoda, N. Photo-Initiated Addition of Diphenyl Ditelluride to Acetylenes. J. Chem. Soc. Chem. Commun. 1991, 24, 1748–1750. [Google Scholar] [CrossRef]
- Ogawa, A.; Yokoyama, H.; Yokoyama, K.; Masawaki, T.; Kambe, N.; Sonoda, N. Photoinitiated Addition of Diphenyl Diselenide to Acetylene. J. Org. Chem. 1991, 56, 5721–5723. [Google Scholar] [CrossRef]
- Ogawa, A.; Yokoyama, K.; Yokoyama, H.; Sekiguchi, M.; Kambe, N.; Sonoda, N. Photo-Initiated Addition of Diphenyl Diselenide to Allenes. Tetrahedron Lett. 1990, 31, 5931–5934. [Google Scholar] [CrossRef]
- Ogawa, A.; Yokoyama, K.; Obayashi, R.; Han, L.B.; Kambe, N.; Sonoda, N. Photo-Induced Ditelluration of Acetylenes with Diphenyl Ditelluride. Tetrahedron 1993, 49, 1177–1188. [Google Scholar] [CrossRef]
- Yamamoto, Y.; Tanaka, R.; Kodama, S.; Nomoto, A.; Ogawa, A. Photoinduced Bisphosphination of Alkynes with Phosphorus Interelement Compounds and Its Application to Double-Bond Isomerization. Molecules 2022, 27, 1284. [Google Scholar] [CrossRef]
- Ito, O. Kinetic Study for Reactions of Phenylseleno Radical with Vinyl Monomers. J. Am. Chem. Soc. 1983, 105, 850–853. [Google Scholar] [CrossRef]
- Ogawa, A.; Tanaka, H.; Yokoyama, H.; Obayashi, R.; Yokoyama, K.; Sonoda, N. A Highly Selective Thioselenation of Olefins Using Disulfide-Diselenide Mixed System. J. Org. Chem. 1992, 57, 111–115. [Google Scholar] [CrossRef]
- Russell, G.A.; Tashtoush, H. Free-Radical Chain-Substitution Reactions of Alkylmercury Halides. J. Am. Chem. Soc. 1983, 105, 1398–1399. [Google Scholar] [CrossRef]
- Tsuchii, K.; Tsuboi, Y.; Kawaguchi, S.I.; Takahashi, J.; Sonoda, N.; Nomoto, A.; Ogawa, A. Highly Selective Double Chalcogenation of Isocyanides with Disulfide-Diselenide Mixed Systems. J. Org. Chem. 2007, 72, 415–423. [Google Scholar] [CrossRef]
- Ogawa, A.; Obayashi, R.; Ine, H.; Tsuboi, Y.; Sonoda, N.; Hirao, T. Highly Regioselective Thioselenation of Acetylenes by Using a (PhS)2-(PhSe)2 Binary System. J. Org. Chem. 1998, 63, 881–884. [Google Scholar] [CrossRef]
- Ogawa, A.; Obayashi, R.; Doi, M.; Sonoda, N.; Hirao, T. A Novel Photoinduced Thioselenation of Allenes by Use of a Disulfide-Diselenide Binary System. J. Org. Chem. 1998, 63, 4277–4281. [Google Scholar] [CrossRef]
- Ogawa, A.; Ogawa, I.; Obayashi, R.; Umezu, K.; Doi, M.; Hirao, T. Highly Selective Thioselenation of Vinylcyclopropanes with a (PhS)2-(PhSe)2 Binary System and Its Application to Thiotelluration. J. Org. Chem. 1999, 64, 86–92. [Google Scholar] [CrossRef]
- Ogawa, A.; Obayashi, R.; Sonoda, N.; Hirao, T. Diphenyl Diselenide-Assisted Dithiolation of 1,3-Dienes with Diphenyl Disulfide upon Irradiation with near-UV Light. Tetrahedron Lett. 1998, 39, 1577–1578. [Google Scholar] [CrossRef]
- Khursan, S.L.; Mikhailov, D.A.; Yanborisov, V.M.; Borisov, D.I. AM1 calculations of bond dissociation energies. Allylic and benzylic C-H bonds. React. Kinet. Catal. Lett. 1997, 61, 91–95. [Google Scholar] [CrossRef]
- Ruscic, B. Active Thermochemical Tables: Sequential Bond Dissociation Enthalpies of Methane, Ethane, and Methanol and the Related Thermochemistry. J. Phys. Chem. A 2015, 119, 7810–7837. [Google Scholar] [CrossRef] [Green Version]
- Chivers, T.; Laitinen, R.S. Tellurium: A Maverick among the Chalcogens. Chem. Soc. Rev. 2015, 44, 1725–1739. [Google Scholar] [CrossRef]
- Yi, M.-J.; Zhang, H.-X.; Xiao, T.-F.; Zhang, J.-H.; Feng, Z.-T.; Wei, L.-P.; Xu, G.-Q.; Xu, P.-F. Photoinduced Metal-Free α-C(sp3)–H Carbamoylation of Saturated Aza-Heterocycles via Rationally Designed Organic Photocatalyst. ACS Catal. 2021, 11, 3466–3472. [Google Scholar] [CrossRef]
- Yoshida, J.; Nakatani, S.; Isoe, S. Electroinitiated oxygenation of alkenyl sulfides and alkynes in the presence of thiophenol. J. Org. Chem. 1993, 58, 4855–4865. [Google Scholar] [CrossRef]
- Wei, C.; He, Y.; Wang, J.; Ye, X.; Wojtas, L.; Shi, X. Hexafluoroisopropanol-Promoted Disulfidation and Diselenation of Alkyne, Alkene, and Allene. Org. Lett. 2020, 22, 5462–5465. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||
|---|---|---|---|---|---|---|
| Entry | (PhS)2 (Equiv.) | (PhSe)2 (mol%) | Solvent (mL) | Time (h) | Yield (%) a | |
| 4 [trans/cis] | 5 | |||||
| 1 | 0.5 | 30 | neat | 20 | 29 [79:21] | 4 |
| 2 | 0.5 | 30 | CDCl3 (0.10) | 20 | 20 [75:25] | 2 |
| 3 | 0.5 | 30 | neat | 100 | 8 | N. D. |
| 4 | 1.0 | 100 | neat | 20 | 41 [93:7] | 3 |
| 5 b | 1.0 | 100 | neat | 50 | 44 [89:11] | trace |
| 6 | 1.0 | - | neat | 20 | 6 [83:17] | N. D. |
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | (PhSe)2 (mol%) | Light Source | CDCl3 (mL) | Time (h) | Yield (%) a | Recovery 1j (%) a | |
| 6a | 7a | ||||||
| 1 | 30 | 100 W Xe lamp | 0.5 | 11 | 11 | 21 | 33 |
| 2 | 30 | 500 W Xe lamp | 0.1 | 10 | 30 | N. D. | N. D. |
| 3 | 100 | 500 W Xe lamp | 0.1 | 10 | 35 | N. D. | 4 |
| 4 | 200 | 500 W Xe lamp | 0.1 | 10 | 39 | 23 | 13 |
 | |||||
|---|---|---|---|---|---|
| Entry | (PhS)2 (Equiv.) | (PhTe)2 (mol%) | Time (h) | Yield (%) a | |
| 8 | 9 [endo/exo] | ||||
| 1 b | 2.0 | 30 | 13 | 2 | 30 [23:77] |
| 2 | 1.0 | 30 | 26 | 4 | 40 [20:80] |
| 3 | 2.0 | 100 | 26 | 3 | 60 [25:75] |
| 4 | 2.0 | 30 | 50 | 10 | 39 [37:63] |
| 5 c | 2.0 | 30 | 26 | 7 | 40 [21:79] |
 | |||||
|---|---|---|---|---|---|
| Entry | CDCl3 (mL) | Time (h) | Yield (%) a | Recovery of 10a (%) a | |
| 11a | 12a | ||||
| 1 | 0.50 | 50 | 45 | trace | 50 |
| 2 | 0.10 | 24 | 61 | trace | 31 |
| 3 | 0.10 | 50 | 81 (70) | trace | 13 |
| 4 b | 0.10 | 50 | 11 | N. D. | 85 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamamoto, Y.; Chen, Q.; Ogawa, A. Diphenyl Diselenide-Assisted Radical Addition Reaction of Diphenyl Disulfide to Unsaturated Bonds upon Photoirradiation. Molecules 2023, 28, 2450. https://doi.org/10.3390/molecules28062450
Yamamoto Y, Chen Q, Ogawa A. Diphenyl Diselenide-Assisted Radical Addition Reaction of Diphenyl Disulfide to Unsaturated Bonds upon Photoirradiation. Molecules. 2023; 28(6):2450. https://doi.org/10.3390/molecules28062450
Chicago/Turabian StyleYamamoto, Yuki, Qiqi Chen, and Akiya Ogawa. 2023. "Diphenyl Diselenide-Assisted Radical Addition Reaction of Diphenyl Disulfide to Unsaturated Bonds upon Photoirradiation" Molecules 28, no. 6: 2450. https://doi.org/10.3390/molecules28062450
APA StyleYamamoto, Y., Chen, Q., & Ogawa, A. (2023). Diphenyl Diselenide-Assisted Radical Addition Reaction of Diphenyl Disulfide to Unsaturated Bonds upon Photoirradiation. Molecules, 28(6), 2450. https://doi.org/10.3390/molecules28062450