
Enhanced Tensile Properties, Biostability, and Biocompatibility of Siloxane–Cross-Linked Polyurethane Containing Ordered Hard Segments for Durable Implant Application

Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis
2.2. FT-IR Analysis
2.3. XPS Analysis
2.4. XRD Analysis
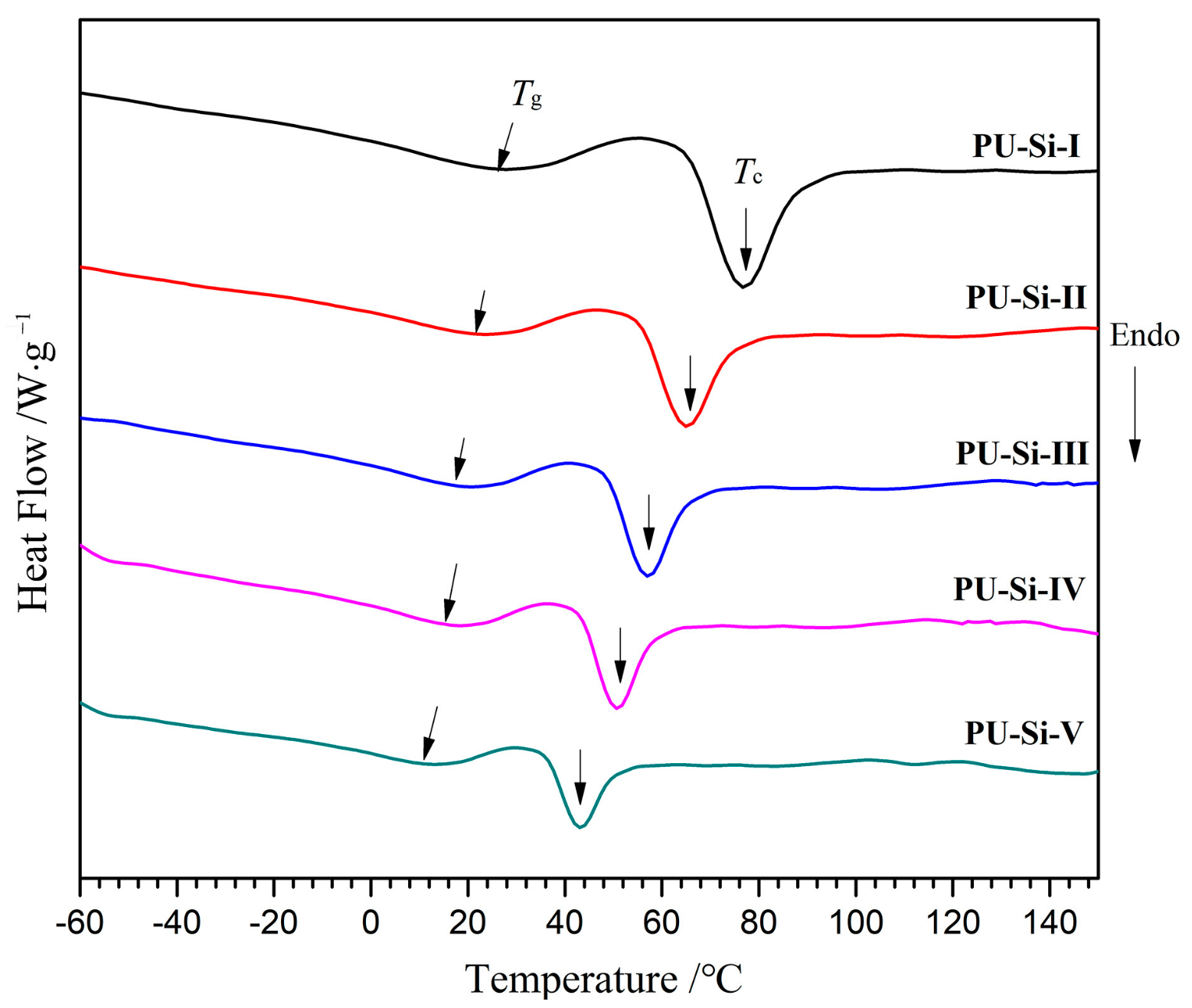
2.5. DSC Analysis
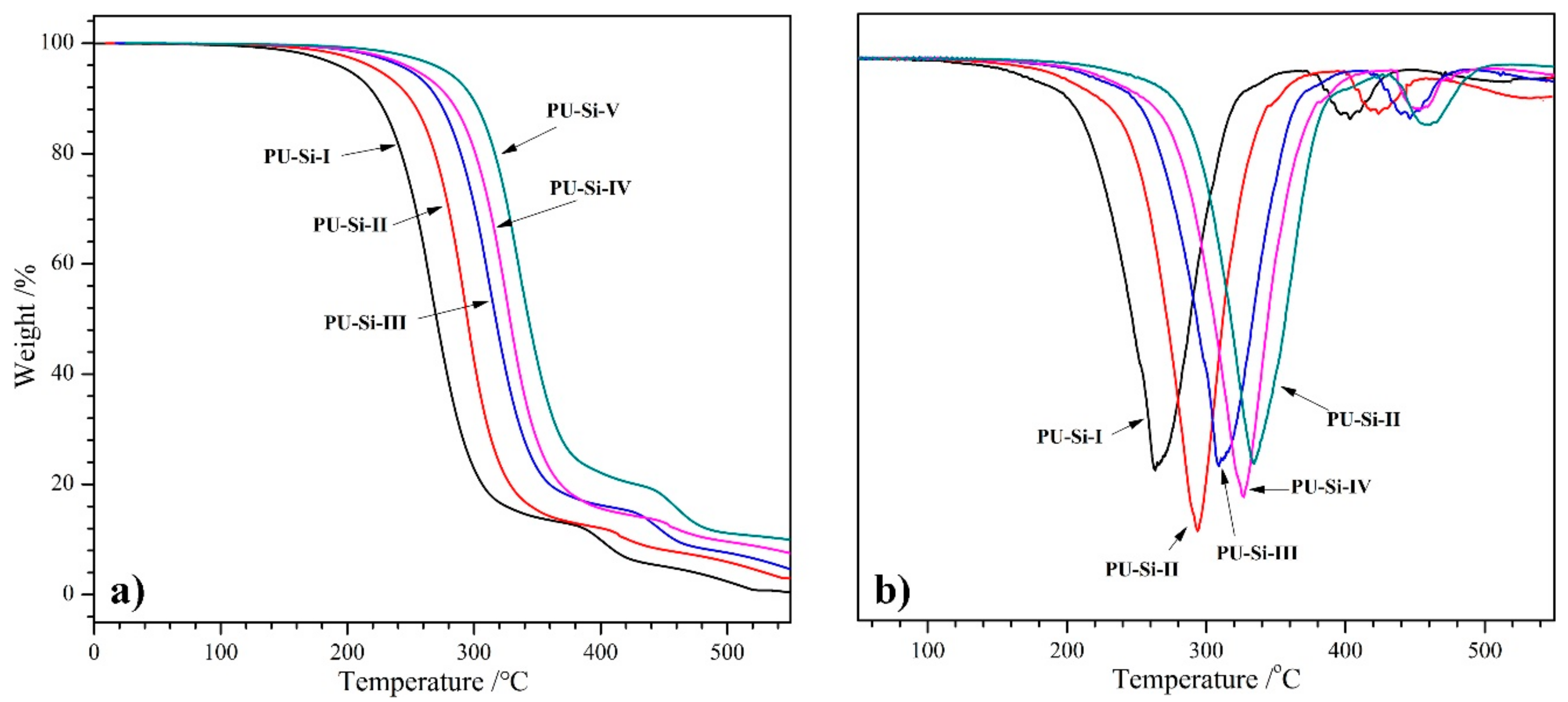
2.6. TGA Analysis
2.7. Tensile Properties
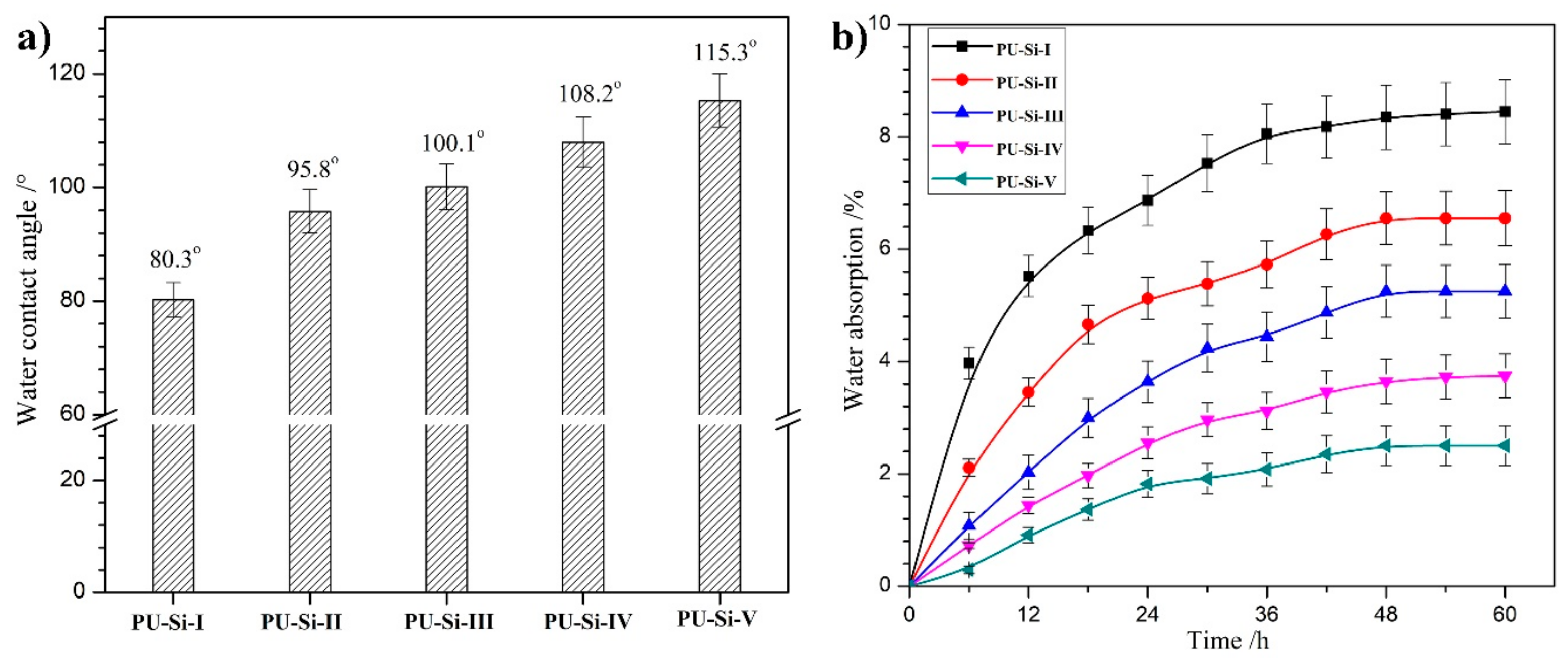
2.8. Surface and Bulk Hydrophilicity
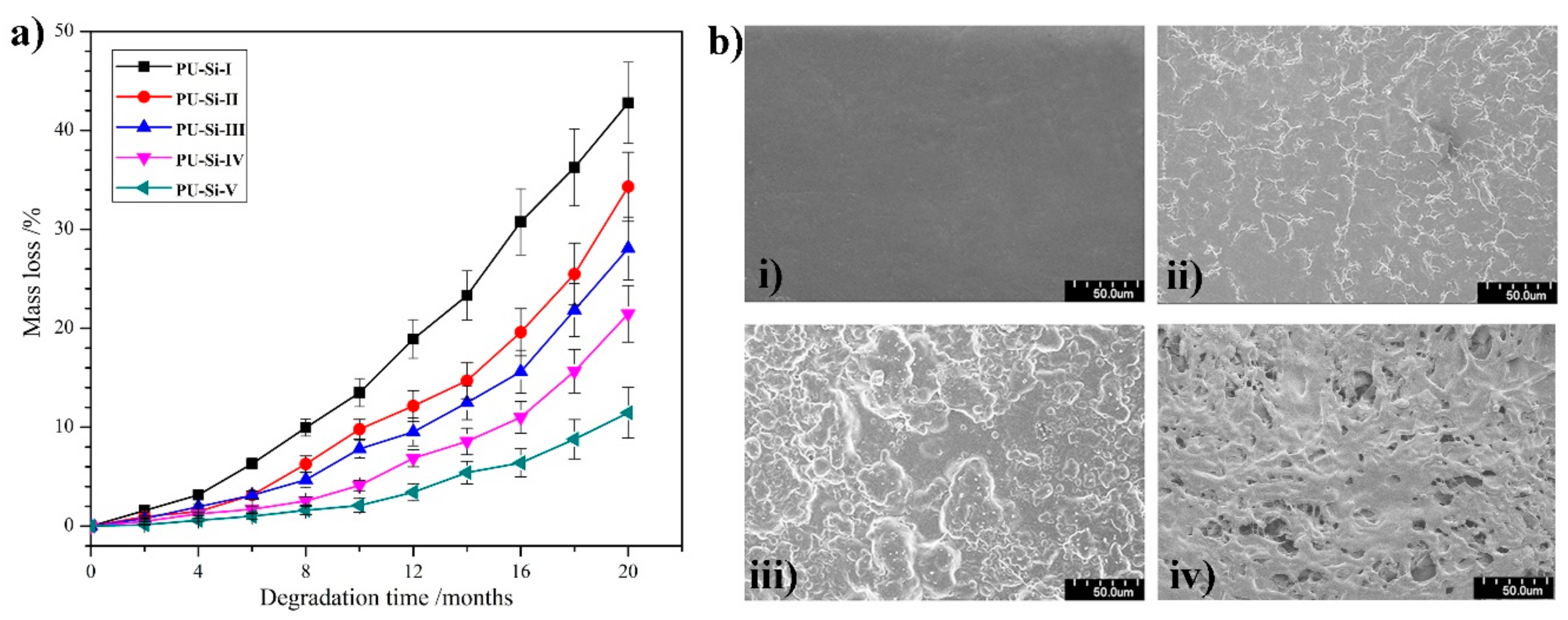
2.9. In Vitro Degradation
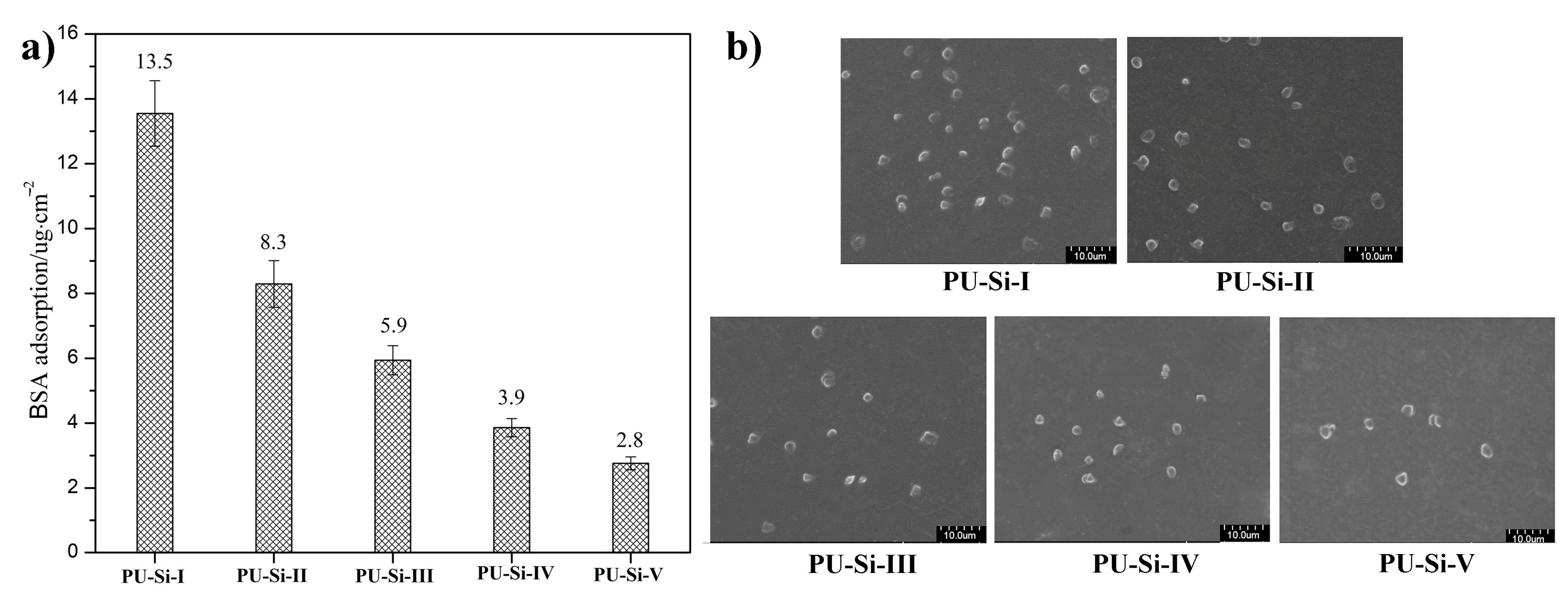
2.10. Surface Biocompatibility
3. Materials and Methods
3.1. Materials
3.2. Preparation of PU–Si and PU–Si Films
3.3. Characterization
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Joshi, M.; Adak, B.; Butola, B.S. Polyurethane Nanocomposite Based Gas Barrier Films, Membranes and Coatings: A Review on Synthesis, Characterization and Potential Applications. Prog. Mater. Sci. 2018, 97, 230–282. [Google Scholar] [CrossRef]
- Akindoyo, J.O.; Beg, M.; Ghazali, S.; Islam, M.R.; Jeyaratnam, N.; Yuvaraj, A.R. Polyurethane Types, Synthesis and Applications—A Review. RSC Adv. 2016, 6, 114453–114482. [Google Scholar] [CrossRef] [Green Version]
- Guo, B.; Glavas, L.; Albertsson, A.C. Biodegradable and Electrically Conducting Polymers for Biomedical Applications. Prog. Polym. Sci. 2013, 38, 1263–1286. [Google Scholar] [CrossRef]
- Janik, H.; Marzec, M. A Review: Fabrication of Porous Polyurethane Scaffolds. Mat. Sci. Eng. C 2015, 48, 586–591. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Liu, Y.; Liu, J. A Facile and Cost-Effective Method to Prepare Biodegradable Poly(Ester Urethane)s with Ordered Aliphatic Hard-Segments for Promising Medical Application as Long-Term Implants. Polymers 2022, 14, 1674. [Google Scholar] [CrossRef]
- Li, D.; Chen, H.; McClung, M.G.; Brash, J.L. Lysine-PEG-Modified Polyurethane as a Fibrinolytic Surface: Effect of PEG Chain Length on Protein Interactions, Platelet Interactions and Clot Lysis. Acta Biomater. 2009, 5, 1864–1871. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, Z.; Gao, Y.; Gao, W.; Hou, Z.; Zhu, Y. Facile Method for Surface-Grafted Chitooligosaccharide on Medical Segmented Poly (Ester-Urethane) Film to Improve Surface Biocompatibility. Membranes 2021, 11, 37. [Google Scholar] [CrossRef]
- Liu, X.; Xia, Y.; Liu, L.; Zhang, D.; Hou, Z. Synthesis of a Novel Biomedical Poly(Ester Urethane) Based on Aliphatic Uniform-Size Diisocyanate and the Blood Compatibility of PEG-Grafted Surfaces. J. Biomater. Appl. 2018, 32, 1329–1342. [Google Scholar] [CrossRef]
- Sun, W.; Liu, W.; Wu, Z.; Chen, H. Chemical Surface Modification of Polymeric Biomaterials for Biomedical Applications. Macromol. Rapid Comm. 2020, 41, 1900430. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, S.; Ding, K.; Zhang, Y.; Ding, X.; Mi, J. Quaternary Tannic Acid with Improved Leachability and Biocompatibility for Antibacterial Medical Thermoplastic Polyurethane Catheters. J. Mater. Chem. B 2021, 9, 4746–4762. [Google Scholar] [CrossRef]
- Najafabadi, S.A.A.; Keshvari, H.; Ganji, Y.; Tahriri, M.; Ashun, M. Chitosan/Heparin Surface Modified Polyacrylic Acid Grafted Polyurethane Film by Two Step Plasma Treatment. Sur. Eng. 2012, 28, 710–714. [Google Scholar] [CrossRef]
- Adipurnama, I.; Yang, M.C.; Ciach, T.; Butruk-Raszeja, B. Surface Modification and Endothelialization of Polyurethane for Vascular Tissue Engineering Applications: A Review. Biomater. Sci. 2017, 5, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Nemani, S.K.; Annavarapu, R.K.; Mohammadian, B.; Raiyan, A.; Heil, J.; Haque, M.A.; Abdelaal, A.; Sojoudi, H. Surface Modification of Polymers: Methods and Applications. Adv. Mater. 2018, 5, 1801247. [Google Scholar] [CrossRef]
- Hou, Z.; Xu, J.; Teng, J.; Jia, Q.; Wang, X. Facile Preparation of Medical Segmented Poly(Ester-Urethane) Containing Uniformly Sized Hard Segments and Phosphorylcholine Groups for Improved Hemocompatibility. Mat. Sci. Eng. C 2020, 109, 110571. [Google Scholar] [CrossRef] [PubMed]
- Abbasi, F.; Mirzadeh, H.; Katbab, A.A. Modification of Polysiloxane Polymers for Biomedical Applications: A Review. Polym. Int. 2001, 50, 1279–1287. [Google Scholar] [CrossRef]
- Grushevenko, E.A.; Borisov, I.L.; Volkov, A.V. High-Selectivity Polysiloxane Membranes for Gases and Liquids Separation (A Review). Petrol. Chem. 2021, 61, 959–976. [Google Scholar] [CrossRef]
- Blanco, I. Polysiloxanes in Theranostics and Drug Delivery: A Review. Polymers 2018, 10, 755. [Google Scholar] [CrossRef] [Green Version]
- Lejars, M.; Margaillan, A.; Bressy, C. Fouling Release Coatings: A Nontoxic Alternative to Biocidal Antifouling Coatings. Chem. Rev. 2012, 112, 4347–4390. [Google Scholar] [CrossRef]
- Galhenage, T.P.; Hoffman, D.; Silbert, S.D.; Stafslien, S.J.; Daniels, J.; Miljkovic, T.; Finlay, J.A.; Franco, S.C.; Clare, A.S.; Nedved, B.T.; et al. Fouling-Release Performance of Silicone Oil-Modified Siloxane-Polyurethane Coatings. ACS Appl. Mater. Interfaces 2016, 8, 29025–29036. [Google Scholar] [CrossRef] [Green Version]
- Gunatillake, P.A.; Dandeniyage, L.S.; Adhikari, R.; Bown, M.; Shanks, R.; Adhikari, B. Advancements in the Development of Biostable Polyurethanes. Polym. Rev. 2019, 59, 391–417. [Google Scholar] [CrossRef]
- Sun, Z.; Wen, J.; Xu, C.; Wang, W.; Fan, H.; Chen, Y.; Xiang, J. Modification of Two-Package Polyurethane by Polyethersiloxanediol for Non-Polar Substrate Coating. Chem. Phys. Lett. 2021, 779, 138878. [Google Scholar] [CrossRef]
- Liu, J.; Pan, Z.; Gao, Y. Study on the Morphology Structure and Properties of Aqueous Polyurethane Modified by Poly(Dimethyl Siloxane). J. Appl. Polym. Sci. 2007, 105, 3037–3046. [Google Scholar]
- Teng, J.; Wang, X.; Xu, J.; Hu, T.; Hou, Z.; Liu, Y. Facile Method for Covalent-Bonding Coating of Crosslinked Silicone Layer onto Poly(Ester-Urethane) Surface to Improve Tensile Properties and Hemocompatibility. Prog. Org. Coat. 2021, 152, 106111. [Google Scholar] [CrossRef]
- Zhao, J.; Xu, R.; Luo, G.; Wu, J.; Xia, H. Self-healing poly (siloxane-urethane) elastomers with remoldability, shape memory and biocompatibility. Polym. Chem. 2016, 7, 7278–7286. [Google Scholar] [CrossRef]
- Verdolotti, L.; Oliviero, M.; Lavorgna, M.; Santillo, C.; Tallia, F.; Iannace, S.; Chen, S.; Jones, J.R. “Aerogel-like” polysiloxane-polyurethane hybrid foams with enhanced mechanical and thermal-insulating properties. Compos. Sci. Technol. 2021, 213, 108917. [Google Scholar] [CrossRef]
- Wu, F.; Pickett, K.; Panchal, A.; Liu, M.; Lvov, Y. Superhydrophobic polyurethane foam coated with polysiloxane-modified clay nanotubes for efficient and recyclable oil absorption. ACS Appl. Mater. Interfaces 2019, 11, 25445–25456. [Google Scholar] [CrossRef] [PubMed]
- Shan, S.; Lin, Y.; Zhang, A. Stretchable, robust and reprocessable poly (siloxane-urethanes) elastomers based on exchangeable aromatic disulfides. Polymer 2021, 221, 123588. [Google Scholar] [CrossRef]
- Palencia, M.; Lerma, T.A.; Arrieta, Á.A. Antibacterial and Non-Hemolytic Cationic Polyurethanes with N-Carboxymethyl-N,N,N-Triethylammonium Groups for Bacteremia-Control in Biomedical-Using Materials. Mater. Today Commun. 2020, 22, 100708. [Google Scholar] [CrossRef]
- Szycher, M.; Siciliano, A.A. An Assessment of 2,4 TDA Formation from Surgitek Polyurethane Foam under Simulated Physiological Conditions. J. Biomater. Appl. 1991, 5, 323–336. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, C.; Zhang, W.; Zhang, H.; Hou, Z. Synthesis and Properties of Biodegradable Poly(Ester-Urethane)s Based on Poly(ε-caprolactone) and Aliphatic Diurethane Diisocyanate for Long-Term Implant Application: Effect of Uniform-Size Hard Segment Content. J. Biomat. Sci. Polym. E. 2019, 30, 1212–1226. [Google Scholar] [CrossRef]
- Meera, K.M.S.; Sankar, R.M.; Jaisankar, S.N.; Mandal, A.B. Physicochemical Studies on Polyurethane/Siloxane Cross-Linked Films for Hydrophobic Surfaces by the Sol−Gel Process. J. Phys. Chem. B 2013, 117, 2682–2694. [Google Scholar] [CrossRef] [PubMed]
- Galhenage, T.P.; Webster, D.C.; Moreira, A.M.; Burgett, R.J.; Stafslien, S.J.; Vanderwal, L.; Finlay, J.A.; Franco, S.C.; Clare, A.S. Poly(Ethylene) Glycol-Modified, Amphiphilic, Siloxane–Polyurethane Coatings and Their Performance as Fouling-Release Surfaces. J. Coat. Technol. Res. 2017, 14, 307–322. [Google Scholar] [CrossRef] [Green Version]
- Nourmohammadi, J.; Ghaee, A.; Liavali, S.H. Preparation and Characterization of Bioactive Composite Scaffolds from Polycaprolactone Nanofibers-Chitosan-Oxidized Starch for Bone Regeneration. Carbohyd. Polym. 2016, 138, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Nanda, A.K.; Wicks, D.A.; Madbouly, S.A.; Otaigbe, J.U. Nanostructured Polyurethane/POSS Hybrid Aqueous Dispersions Prepared by Homogeneous Solution Polymerization. Macromolecules 2006, 39, 7037–7043. [Google Scholar] [CrossRef]
- Yang, B.; Yin, S.; Bian, X.; Liu, C.; Liu, X.; Yan, Y.; Zhang, C.; Zhang, H.; Hou, Z. Preparation and Properties of Monomethoxyl Polyethylene Glycol Grafted O-Carboxymethyl Chitosan for Edible, Fresh-Keeping Packaging Materials. Food Packaging Shelf. 2022, 33, 100874. [Google Scholar] [CrossRef]
- Zhao, H.; Hao, T.H.; Hu, G.H.; Shi, D.; Huang, D.; Jiang, T.; Zhang, Q.C. Preparation and Characterization of Polyurethanes with Cross-Linked Siloxane in the Side Chain by Sol-Gel Reactions. Materials 2017, 10, 247. [Google Scholar] [CrossRef] [Green Version]
- Jia, R.; Hui, Z.; Huang, Z.; Liu, X.; Zhao, C.; Wang, D.; Wu, D. Synthesis and Antibacterial Investigation of Cationic Waterborne Polyurethane Containing Siloxane. New J. Chem. 2020, 44, 19759–19768. [Google Scholar] [CrossRef]
- Allauddin, S.; Narayan, R.; Raju, K.V.S.N. Synthesis and Properties of Alkoxysilane Castor Oil and Their Polyurethane/Urea–Silica Hybrid Coating Films. ACS Sustain. Chem. Eng. 2013, 1, 910–918. [Google Scholar] [CrossRef]
- Abraham, G.A.; Frontini, P.M.; Cuadrado, T.R. Molding of Biomedical Segmented Polyurethane Delamination Events and Stretching Behavior. J. Appl. Polym. Sci. 2015, 69, 2159–2167. [Google Scholar] [CrossRef]
- Wu, G.; Liu, D.; Chen, J.; Liu, G.; Kong, Z. Preparation and Properties of Super Hydrophobic Films from Siloxane-Modified Two-Component Waterborne Polyurethane and Hydrophobic Nano SiO2. Prog. Org. Coat. 2019, 127, 80–87. [Google Scholar] [CrossRef]
- Zhao, X.; Li, Y.; Li, B.; Hu, T.; Yang, Y.; Li, L.; Zhang, J. Environmentally Benign and Durable Superhydrophobic Coatings Based on SiO2 Nanoparticles and Silanes. J. Colloid Interface Sci. 2019, 542, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Zia, K.M.; Ahmad, A.; Anjum, S.; Zuber, M.; Anjum, M.N. Synthesis and Characterization of Siloxane-Based Polyurethane Elastomers Using Hexamethylene Diisocyanate. J. Elastom. Plast. 2015, 47, 625–635. [Google Scholar] [CrossRef]
- Benda, J.; Stafslien, S.; Vanderwal, L.; Finlay, J.A.; Clare, A.S.; Webster, D.C. Surface Modifying Amphiphilic Additives and Their Effect on the Fouling-Release Performance of Siloxane-Polyurethane Coatings. Biofouling 2021, 37, 309–326. [Google Scholar] [CrossRef] [PubMed]
- Rahimi, A.; Stafslien, S.J.; Vanderwal, L.; Finlay, J.A.; Clare, A.S.; Webster, D.C. Amphiphilic Zwitterionic-PDMS-Based Surface-Modifying Additives to Tune Fouling-Release of Siloxane-Polyurethane Marine Coatings. Prog. Org. Coat. 2020, 149, 105931. [Google Scholar] [CrossRef]
- Shih, M.F.; Shau, M.D.; Hsieh, C.C.; Cherng, J.Y. Synthesis and Evaluation of Poly(Hexamethylene-Urethane) and PEG-Poly(Hexamethylene-Urethane) and Their Cholesteryl Oleyl Carbonate Composites for Human Blood Biocompatibility. Molecules 2011, 16, 8181–8197. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Film Samples | Tg/°C | Tc/°C | ΔHm/J·g−1 |
|---|---|---|---|
| PU–Si–I | 26.3 | 76.8 | 19.1 |
| PU–Si–II | 22.0 | 65.3 | 15.7 |
| PU–Si–III | 17.8 | 57.0 | 13.6 |
| PU–Si–IV | 15.1 | 50.8 | 11.2 |
| PU–Si–V | 12.2 | 42.9 | 8.9 |
| Film Samples | T5%/°C | Tmax–1/°C | Tmax–2/°C | Wr/% |
|---|---|---|---|---|
| PU–Si–I | 201 | 263 | 404 | 0.8 |
| PU–Si–II | 226 | 293 | 423 | 3.1 |
| PU–Si–III | 248 | 309 | 446 | 4.9 |
| PU–Si–IV | 255 | 326 | 454 | 7.5 |
| PU–Si–V | 281 | 334 | 460 | 10.1 |
| Film Samples | UTS/MPa | UE/% | YM/MPa | FT/MJ·m−3 |
|---|---|---|---|---|
| PU–Si–I | 29.8 ± 1.9 | 1064 ± 85 | 23.2 | 19.1 |
| PU–Si–II | 33.7 ± 2.2 | 944 ± 68 | 41.7 | 21.2 |
| PU–Si–III | 38.4 ± 2.7 | 797 ± 53 | 50.6 | 21.6 |
| PU–Si–IV | 49.2 ± 3.3 | 633 ± 52 | 57.2 | 19.7 |
| PU–Si–V | 54.7 ± 3.8 | 475 ± 31 | 70.8 | 16.9 |
| Film samples | PCL/mmol | HBH/mmol | APTS/mmol | HBH /wt% | APTS/wt% |
|---|---|---|---|---|---|
| PU–Si–I | 4.0 | 4.0 | 0 | 17.5 | 0 |
| PU–Si–II | 4.0 | 5.0 | 2.0 | 20.2 | 4.2 |
| PU–Si–III | 4.0 | 6.0 | 4.0 | 22.4 | 7.7 |
| PU–Si–IV | 4.0 | 7.0 | 6.0 | 24.2 | 10.8 |
| PU–Si–V | 4.0 | 8.0 | 4.0 | 25.9 | 13.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, X.; Jia, H.; Fu, W.; Li, M.; Pan, Y. Enhanced Tensile Properties, Biostability, and Biocompatibility of Siloxane–Cross-Linked Polyurethane Containing Ordered Hard Segments for Durable Implant Application. Molecules 2023, 28, 2464. https://doi.org/10.3390/molecules28062464
Wu X, Jia H, Fu W, Li M, Pan Y. Enhanced Tensile Properties, Biostability, and Biocompatibility of Siloxane–Cross-Linked Polyurethane Containing Ordered Hard Segments for Durable Implant Application. Molecules. 2023; 28(6):2464. https://doi.org/10.3390/molecules28062464
Chicago/Turabian StyleWu, Xiaofei, Hanxiao Jia, Wenshuo Fu, Meng Li, and Yitong Pan. 2023. "Enhanced Tensile Properties, Biostability, and Biocompatibility of Siloxane–Cross-Linked Polyurethane Containing Ordered Hard Segments for Durable Implant Application" Molecules 28, no. 6: 2464. https://doi.org/10.3390/molecules28062464
APA StyleWu, X., Jia, H., Fu, W., Li, M., & Pan, Y. (2023). Enhanced Tensile Properties, Biostability, and Biocompatibility of Siloxane–Cross-Linked Polyurethane Containing Ordered Hard Segments for Durable Implant Application. Molecules, 28(6), 2464. https://doi.org/10.3390/molecules28062464