
Cytotoxicity Evaluation of Unmodified Paddlewheel Dirhodium(II,II)-Acetate/-Formamidinate Complexes and Their Axially Modified Low-Valent Metallodendrimers

 ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Synthesis and Characterization
2.2. In Vitro Cytotoxicity Measurements
3. Materials and Methods
3.1. General Information
3.2. Instruments
3.3. Synthetic Procedure for Trimeric Dirhodium(II,II) Metallodendrimers
3.3.1. Tris-(dirhodium(II,II) tetraacetate) Metallodendrimer (8)
3.3.2. Tris-(dirhodium(II,II) tetrakis(di-p-tolylformamidinate)) Metallodendrimer (9)
3.3.3. Tris-(dirhodium(II,II) tetrakis(bis-(4-fluorophenyl)formamidinate)) Metallodendrimer (10)
3.4. Cell Growth Inhibition
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
Abbreviations
| DCM | Dichloromethane |
| DMSO | Dimethylsulfoxide |
| DNA | Deoxyribonucleic acid |
| HR-ESI-MS | High Resolution Electrospray Ionization Mass Spectrometry |
| MTT | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NMR | Nuclear magnetic resonance |
| IC50 | Concentration required for 50% cell growth inhibition |
References
- Gasser, G.; Metzler-Nolte, N. The potential of organometallic complexes in medicinal chemistry. Curr. Opin. Chem. Biol. 2012, 16, 84–91. [Google Scholar] [CrossRef] [Green Version]
- Bruijnincx, P.C.; Sadler, P.J. New trends for metal complexes with anticancer activity. Curr. Opin. Chem. Biol. 2008, 12, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular mechanisms of cisplatin resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [Green Version]
- Reedijk, J. Platinum Anticancer Coordination Compounds: Study of DNA Binding Inspires New Drug Design. Eur. J. Inorg. Chem. 2009, 10, 1303–1312. [Google Scholar] [CrossRef]
- Yan, Y.K.; Cho, S.E.; Shaffer, K.A.; Rowell, J.E.; Barnes, B.J.; Hall, I.H. Cytotoxicity of rhenium(I) alkoxo and hydroxo carbonyl complexes in murine and human tumor cells. Pharmazie 2000, 55, 307–313. [Google Scholar]
- Groessl, M.; Reisner, E.; Hartinger, C.G.; Eichinger, R.; Semenova, O.; Timerbaev, A.R.; Jakupec, M.A.; Arion, V.B.; Keppler, B.K. Structure−Activity Relationships for NAMI-A-type Complexes (HL)[trans-RuCl4L(S-dmso)ruthenate(III)] (L = Imidazole, Indazole, 1,2,4-Triazole, 4-Amino-1,2,4-triazole, and 1-Methyl-1,2,4-triazole): Aquation, Redox Properties, Protein Binding, and Antiproliferative Activity. J. Med. Chem. 2007, 50, 2185–2193. [Google Scholar]
- Ang, W.H.; Grote, Z.; Scopelliti, R.; Juillerat-Jeanneret, L.; Severin, K.; Dyson, P.J. Organometallic complexes that interconvert between trimeric and monomeric structures as a function of pH and their effect on human cancer and fibroblast cells. J. Organomet. Chem. 2009, 694, 968–972. [Google Scholar] [CrossRef]
- Zhang, C.X.; Lippard, S.J. New metal complexes as potential therapeutics. Curr. Opin. Chem. Biol. 2003, 7, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Govender, P.; Riedel, T.; Dyson, P.J.; Smith, G.S. Regulating the anticancer properties of organometallic dendrimers using pyridylferrocene entities: Synthesis, cytotoxicity and DNA binding studies. Dalton Trans. 2016, 45, 9529–9539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, G.; Garci, A.; Murray, B.S.; Dyson, P.J.; Fabre, G.; Trouillas, P.; Giannini, F.; Furrer, J.; Süss-Fink, G.; Therrien, B. Synthesis, molecular structure, computational study and in vitro anticancer activity of dinuclearthiolato-bridged pentamethylcyclopentadienyl Rh(III) and Ir(III) complexes. Dalton Trans. 2013, 42, 15457–15463. [Google Scholar] [CrossRef]
- Barry, N.P.E.; Sadler, P.J. Dicarba-closo-dodecarborane-containing half-sandwich complexes of ruthenium, osmium, rhodium and iridium: Biological relevance and synthetic strategies. Chem. Soc. Rev. 2012, 41, 3264–3279. [Google Scholar] [CrossRef] [Green Version]
- Truong, D.; Sullivan, M.P.; Tong, K.K.H.; Steel, T.R.; Prause, A.; Lovett, J.H.; Andersen, J.W.; Jamieson, S.M.F.; Harris, H.H.; Ott, I.; et al. Potent Inhibition of Thioredoxin Reductase by the Rh Derivatives of Anticancer M(arene/Cp*)(NHC)Cl2 Complexes. Inorg. Chem. 2020, 59, 3281–3289. [Google Scholar] [CrossRef]
- Katsaros, N.; Anagnostopoulou, A. Rhodium and its compounds as potential agents in cancer treatment. Crit. Rev. Oncol. Hematol. 2002, 42, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Baban, D.F.; Seymour, L.W. Control of vascular permeability. Adv. Drug Deliv. Rev. 1998, 34, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Farrell, N. Polynuclear platinum drugs. Met. Ions Biol. Syst. 2002, 42, 251–296. [Google Scholar]
- Chellan, P.; Land, K.M.; Shokar, A.; Au, A.; An, S.H.; Taylor, D.; Smith, P.J.; Riedel, T.; Dyson, P.J.; Chibale, K.; et al. Synthesis and evaluation of new polynuclear organometallic Ru(II), Rh(III) and Ir(III) pyridyl ester complexes as in vitro antiparasitic and antitumor agents. Dalton Trans. 2014, 43, 513–526. [Google Scholar] [CrossRef] [Green Version]
- Makhubela, B.C.E.; Meyer, M.; Smith, G.S. Evaluation of trimetallic Ru(II)- and Os(II)-Arene complexes as potential anticancer agents. J. Organomet. Chem. 2014, 772–773, 229–241. [Google Scholar] [CrossRef]
- Burgoyne, A.R.; Makhubela, B.C.E.; Meyer, M.; Smith, G.S. Trinuclear Half-Sandwich RuII, RhIII and IrIII Polyester Organometallic Complexes: Synthesis and in vitro Evaluation as Antitumor Agents. Eur. J. Inorg. Chem. 2015, 2015, 1433–1444. [Google Scholar] [CrossRef]
- Burgoyne, A.R.; Kaschula, C.H.; Parker, M.I.; Smith, G.S. Synthesis and anticancer evaluation of mono- and trinuclear half-sandwich rhodium(III) and iridium(III) complexes based on N,O salicylaldiminato-sulfonated scaffolds. J. Organomet. Chem. 2017, 846, 100–104. [Google Scholar] [CrossRef]
- Welsh, A.; Rylands, L.; Arion, V.B.; Prince, S.; Smith, G.S. Synthesis and antiproliferative activity of benzimidazole-based, trinuclear neutral cyclometallated and cationic, N^N-chelated ruthenium(II) complexes. Dalton Trans. 2020, 49, 1143–1156. [Google Scholar] [CrossRef]
- Bear, J.L.; Han, B.; Li, Y.; Ngubane, S.; Van Caemelbecke, E.; Kadish, K.M. Synthesis, spectroscopic properties and electrochemistry of Rh2(ap)4(R) where R = CH3 or C6H5 and ap = 2-anilinopyridinate anion. Polyhedron 2009, 28, 1551–1555. [Google Scholar] [CrossRef]
- Frade, R.F.M.; Candeias, N.R.; Duarte, C.M.M.; Andre, V.; Duarte, M.T.; Gois, P.M.P.; Afonso, C.A.M. New dirhodium complex with activity towards colorectal cancer. Bioorg. Med. Chem. Lett. 2010, 20, 3413. [Google Scholar] [CrossRef] [PubMed]
- Howard, R.A.; Sherwood, E.; Erck, A.; Kimball, A.P.; Bear, J.L. Hydrophobicity of several rhodium(II) carboxylates correlated with their biologic activity. J. Med. Chem. 1977, 20, 943–946. [Google Scholar] [CrossRef]
- Zyngier, S.; Kimura, E.; Najjar, R. Antitumor effects of rhodium (II) citrate in mice bearing Ehrlich tumors. Braz. J. Med. Biol. Res. 1989, 22, 397–401. [Google Scholar] [PubMed]
- Dale, L.D.; Dyson, T.M.; Tocher, D.A.; Tocher, J.H.; Edwards, D.I. Studies on DNA damage and induction of SOS repair by novel multifunctional bioreducible compounds. I. A metronidazole adduct of dirhodium(II) tetraacetate. Anti-Cancer Drug Des. 1989, 4, 295–302. [Google Scholar]
- Erck, A.; Rainen, L.; Whileyman, J.; Chang, I.M.; Kimball, A.P.; Bear, J.L. Studies of rhodium(II) carboxylates as potential antitumor agents. Proc. Soc. Exp. Biol. Med. 1974, 145, 1278–1283. [Google Scholar] [CrossRef]
- Clarke, M.J.; Stubbs, M. Interactions of Metallopharmaceuticals with DNA. In Metal Ions in Biological Systems; Sigel, A., Sigel, H., Eds.; Marcel Dekker: New York, NY, USA, 1996; Volume 32. [Google Scholar]
- Knoll, J.D.; Turro, C. Control and utilization of ruthenium and rhodium metal complex excited states for photoactivated cancer therapy. Coord. Chem. Rev. 2015, 282–283, 110–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chifotides, H.T.; Dunbar, K.R. Interactions of Metal-Metal Bonded Antitumor Active Complexes with DNA fragments and DNA. Acc. Chem. Res. 2005, 38, 146–156. [Google Scholar] [CrossRef]
- Chifotides, H.T.; Koshlap, K.M.; Perez, L.M.; Dunbar, K.R. Unprecedented Head-to-Head Conformers of d(GpG) Bound to the Antitumor Active Compound Tetrakis (µ-carboxylato)dirhodium(II,II). J. Am. Chem. Soc. 2003, 125, 10703–10713. [Google Scholar] [CrossRef]
- Aguirre, J.D.; Angeles-Boza, A.M.; Chouai, A.; Pellois, J.P.; Turro, C.; Dunbar, K.R.J. Live Cell Cytotoxicity Studies: Documentation of the Interactions of Antitumor Active Dirhodium Compounds with Nuclear DNA. Am. Chem. Soc. 2009, 131, 11353–11360. [Google Scholar] [CrossRef]
- Piraino, P.; Tresoldi, G.; Lo Schiavo, S. Interactions of Rh24+ formamidinate complex Rh2(µ-form)2(µ-O2CCF3)2(H2O)2 (form = N,N’-di-ptolylformamidinate anion) with nucleobases and nucleosides. Inorg. Chim. Acta 1993, 203, 101–105. [Google Scholar] [CrossRef]
- Howard, R.A.; Spring, T.; Bear, J. The interaction of rhodium (II) carboxylates with enzymes. Cancer Res. 1976, 36, 4402–4405. [Google Scholar]
- Fimiani, V.; Ainis, T.; Cavallaro, A.; Piraino, P. Antitumor effect of the new rhodium(II) complex: Rh2(Form)2(O2CCF3)2(H2O)2 (Form = N,N’-di-p-tolylformamidinate). J. Chemother. 1990, 2, 319–326. [Google Scholar] [CrossRef] [PubMed]
- de Doncker, S.; Casimiro, A.; Kotze, I.A.; Ngubane, S.; Smith, G.S. Bimetallic paddlewheel-type dirhodium(II,II) acetate and formamidinate complexes: Synthesis, structure, electrochemistry and hydroformylation activity. Inorg. Chem. 2020, 59, 12928–12940. [Google Scholar] [CrossRef] [PubMed]
- Giffard, D.; Fischer-Fodor, E.; Vlad, C.; Achimas-Cadariu, P.; Smith, G.S. Synthesis and antitumour evaluation of mono- and multinuclear [2+1] tricarbonylrhenium(I) complexes. Eur. J. Med. Chem. 2018, 157, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Guoyu, Y.; Ailing, S.; Wenfeng, Z.; Hailin, Z.; Denggao, J. Comparative study of ship-in-a-bottle and anchored heterogenized Mn complexes. Catal. Lett. 2007, 118, 275–279. [Google Scholar] [CrossRef]
- Caminade, A.; Turrin, C.O.; Laurent, R.; Ouali, A.; Delavaux-Nicot, B. Dendrimers—Towards Catalytic, Material and Biomedical Uses; John Wiley & Sons: Toulouse, France, 2011. [Google Scholar]
- Miklásová, N.; Fischer-Fodor, E.; Lönnecke, P.; PerdeSchrepler, M.; Virag, P.; Tatomir, C.; Cernea, V.I.; Hey-Hawkins, E.; Silaghi-Dumitrescu, L. Antiproliferative effect and genotoxicity of novel synthesized palladium complexes with organoarsenic ligands. J. Inorg. Biochem. 2009, 103, 1739–1747. [Google Scholar] [CrossRef]
- Hebling, J.; Bianchi, L.; Basso, F.G.; Scheffel, D.L.; Soares, D.G.; Carrilho, M.R.O.; Pashley, D.H.; Tjäderhane, L.; de Souza Costa, C.A. Cytotoxicity of dimethyl sulfoxide (DMSO) in direct contact with odontoblast-like cells. Dent. Mater. 2015, 31, 399–405. [Google Scholar] [CrossRef] [Green Version]
- Hallas-Potts, A.; Dawson, J.C.; Herrington, C.S. Ovarian cancer cell lines derived from non-serous carcinomas migrate and invade more aggressively than those derived from high-grade serous carcinomas. Sci. Rep. 2019, 9, 5515. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
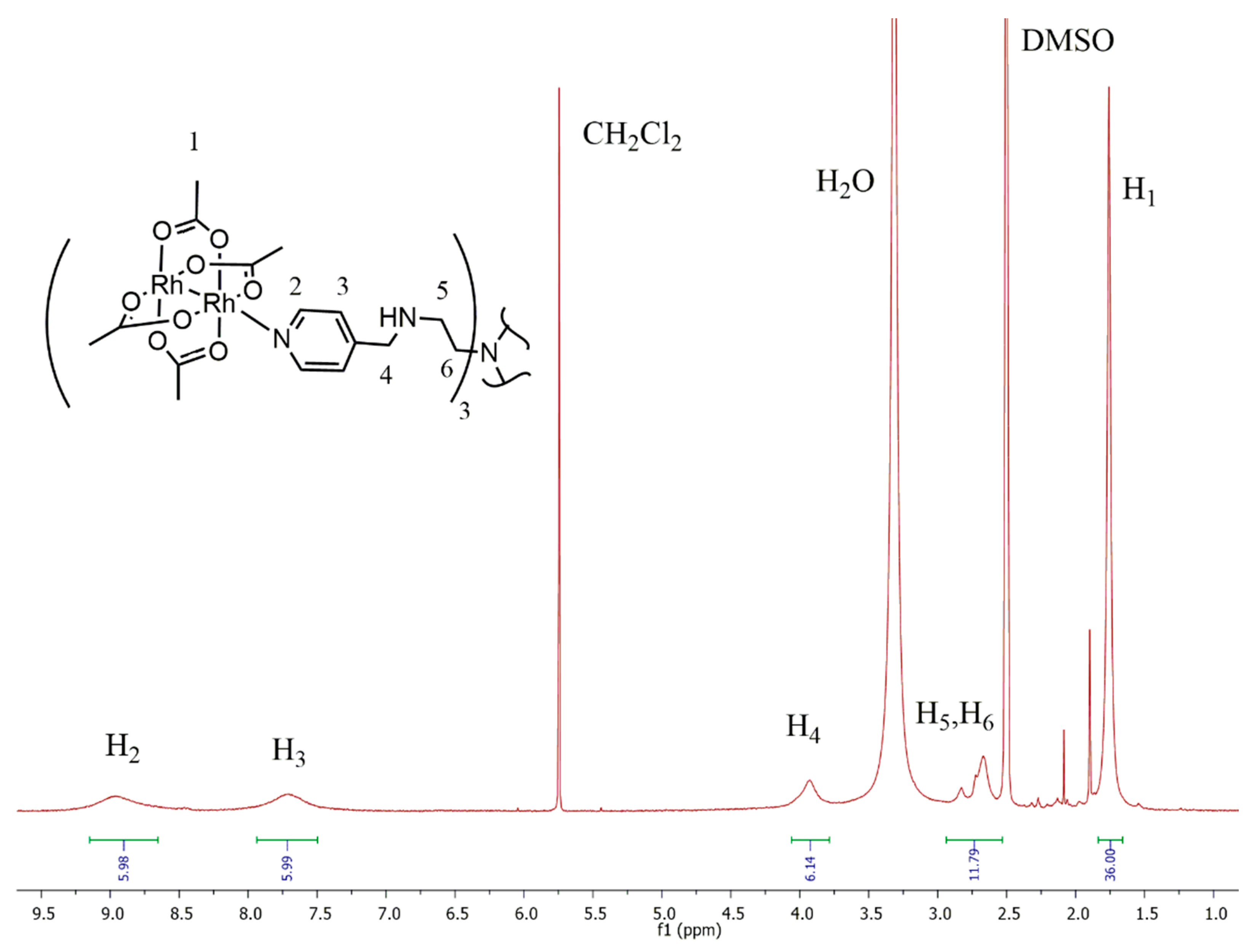{kind=link}
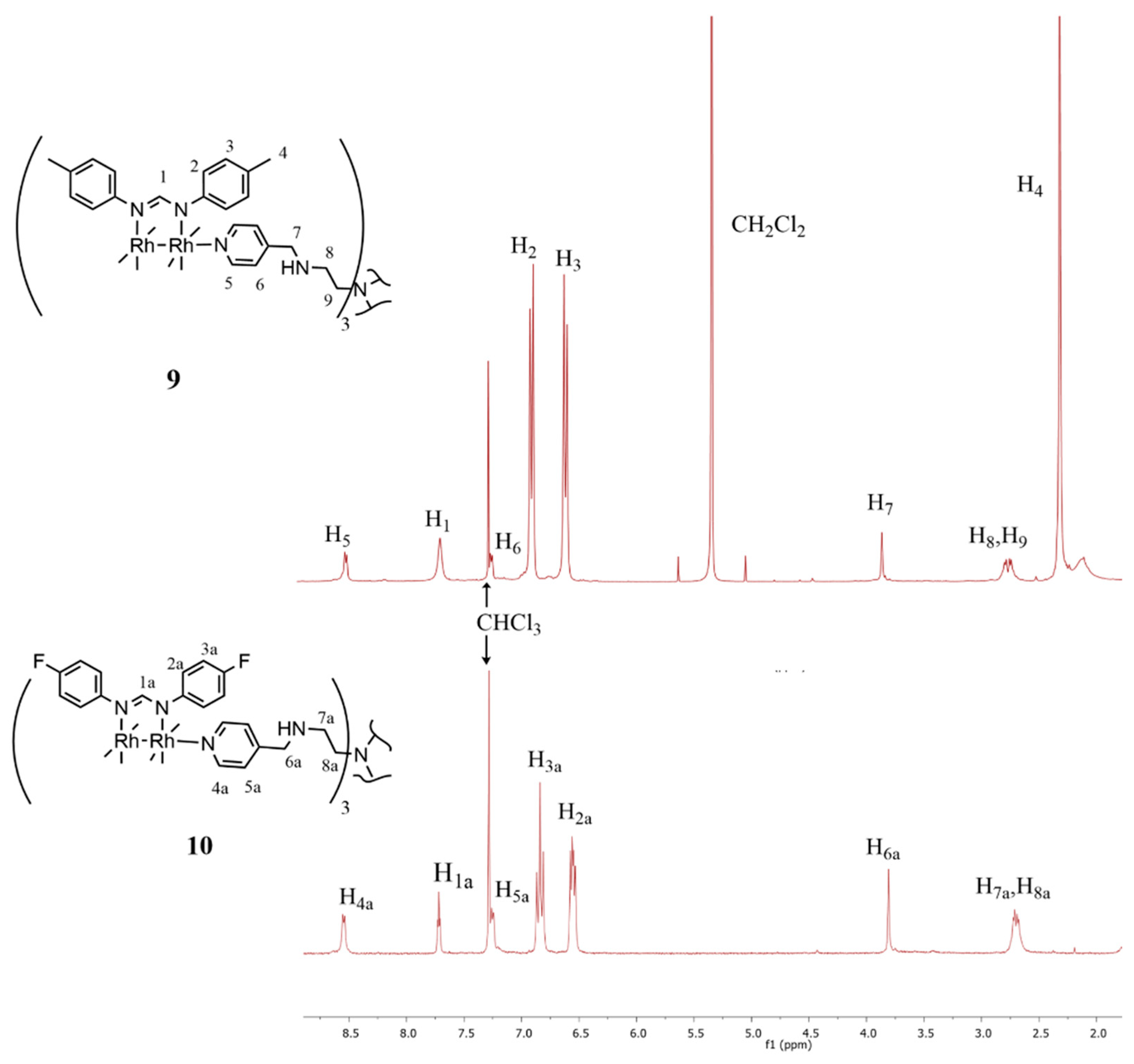{kind=link}
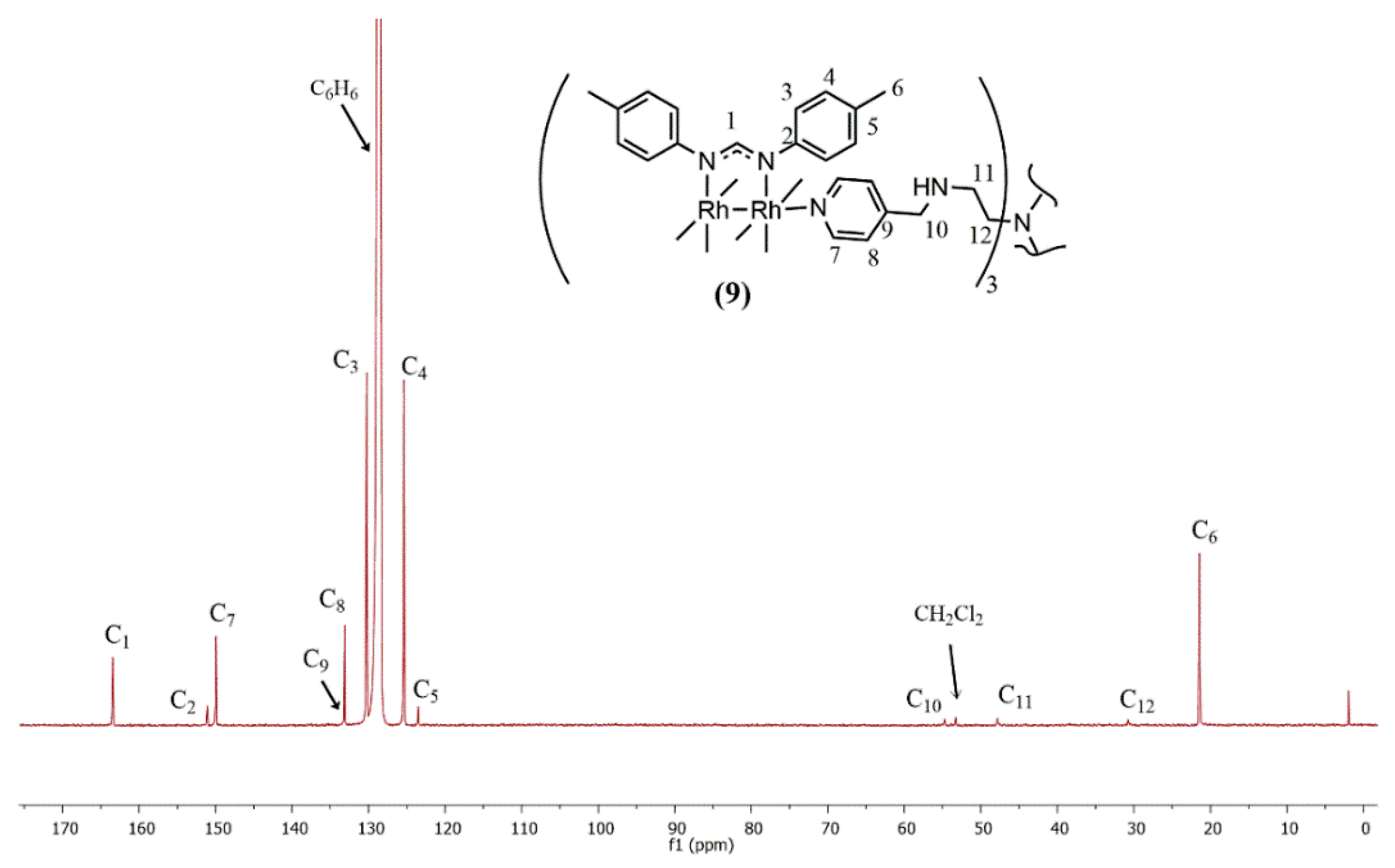{kind=link}
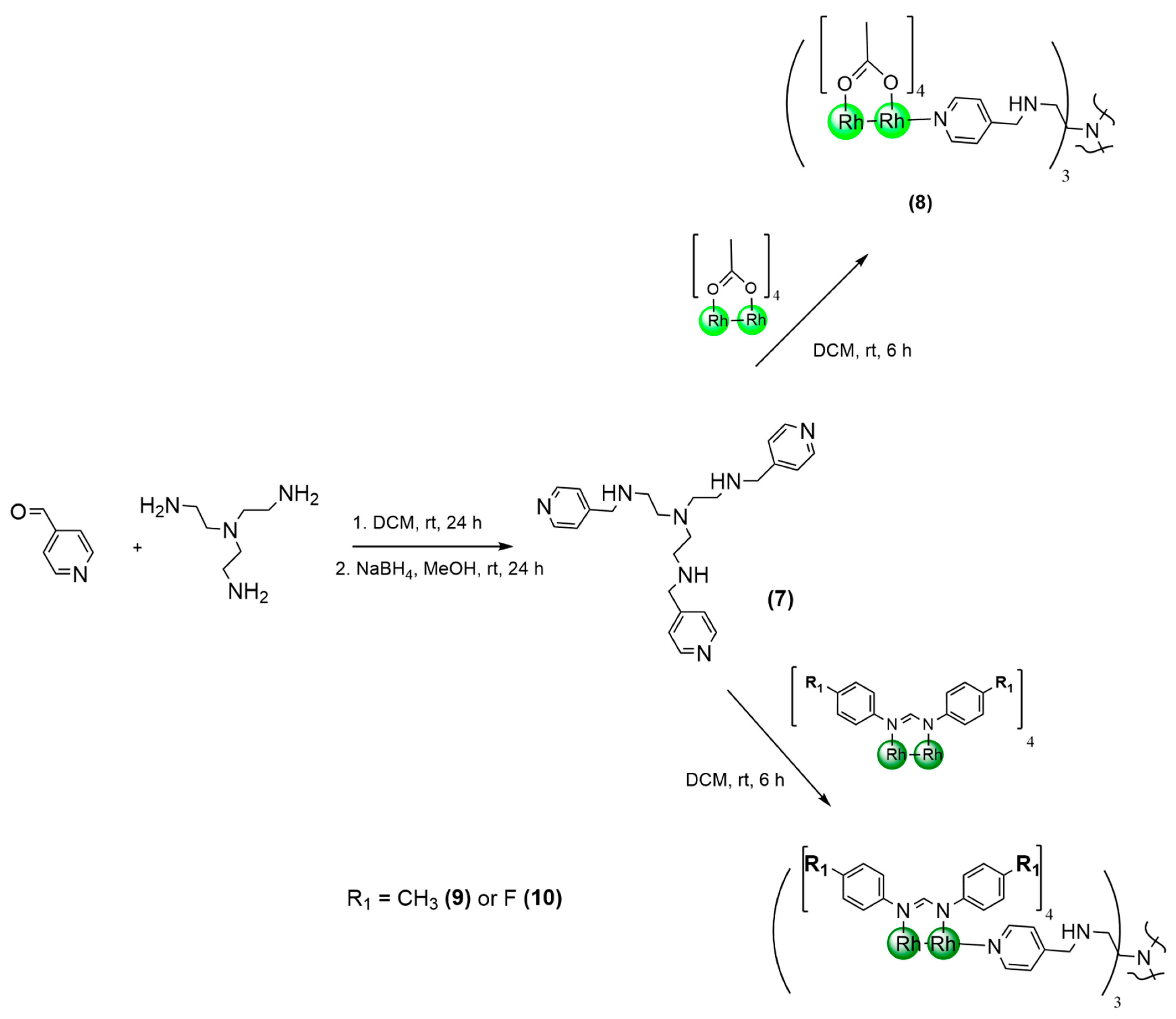{kind=link}
| Compound | n a | Sigmoidal Dose–Response IC50(µM) b | ||
|---|---|---|---|---|
| A2780 | A2780cis | OVCAR-3 | ||
| Cisplatin | 1 | 7.60 ± 1.34 | 80.49 ± 0.74 | 30.57 ± 0.85 |
| Carboplatin | 1 | 53.06 ± 2.52 | 89.37 ± 2.32 | 45.90 ± 0.91 |
| 1 | 0 | * | * | * |
| 2 | 0 | 43.68 ± 0.99 | * | * |
| 3 | 2 | 45.65 ± 0.97 | 43.25 ± 0.94 | * |
| 4 | 2 | 52.57 ± 0.81 | * | 32.22 ± 0.76 |
| 5 | 2 | 36.18 ± 0.97 | 36.24 ± 0.97 | 27.71 ± 1.09 |
| 6 | 2 | 41.17 ± 0.87 | * | 72.21 ± 0.74 |
| 8 | 6 | 64.82 ± 1.23 | * | 69.05 ± 0.52 |
| 9 | 6 | * | * | * |
| 10 | 6 | 97.11 ± 1.02 | * | * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Doncker, S.; Fischer-Fodor, E.; Vlad, C.I.; Achimas-Cadariu, P.; Smith, G.S.; Ngubane, S. Cytotoxicity Evaluation of Unmodified Paddlewheel Dirhodium(II,II)-Acetate/-Formamidinate Complexes and Their Axially Modified Low-Valent Metallodendrimers. Molecules 2023, 28, 2671. https://doi.org/10.3390/molecules28062671
de Doncker S, Fischer-Fodor E, Vlad CI, Achimas-Cadariu P, Smith GS, Ngubane S. Cytotoxicity Evaluation of Unmodified Paddlewheel Dirhodium(II,II)-Acetate/-Formamidinate Complexes and Their Axially Modified Low-Valent Metallodendrimers. Molecules. 2023; 28(6):2671. https://doi.org/10.3390/molecules28062671
Chicago/Turabian Stylede Doncker, Stephen, Eva Fischer-Fodor, Cătălin Ioan Vlad, Patriciu Achimas-Cadariu, Gregory S. Smith, and Siyabonga Ngubane. 2023. "Cytotoxicity Evaluation of Unmodified Paddlewheel Dirhodium(II,II)-Acetate/-Formamidinate Complexes and Their Axially Modified Low-Valent Metallodendrimers" Molecules 28, no. 6: 2671. https://doi.org/10.3390/molecules28062671
APA Stylede Doncker, S., Fischer-Fodor, E., Vlad, C. I., Achimas-Cadariu, P., Smith, G. S., & Ngubane, S. (2023). Cytotoxicity Evaluation of Unmodified Paddlewheel Dirhodium(II,II)-Acetate/-Formamidinate Complexes and Their Axially Modified Low-Valent Metallodendrimers. Molecules, 28(6), 2671. https://doi.org/10.3390/molecules28062671