3.2. Chemical Synthesis and Characterization
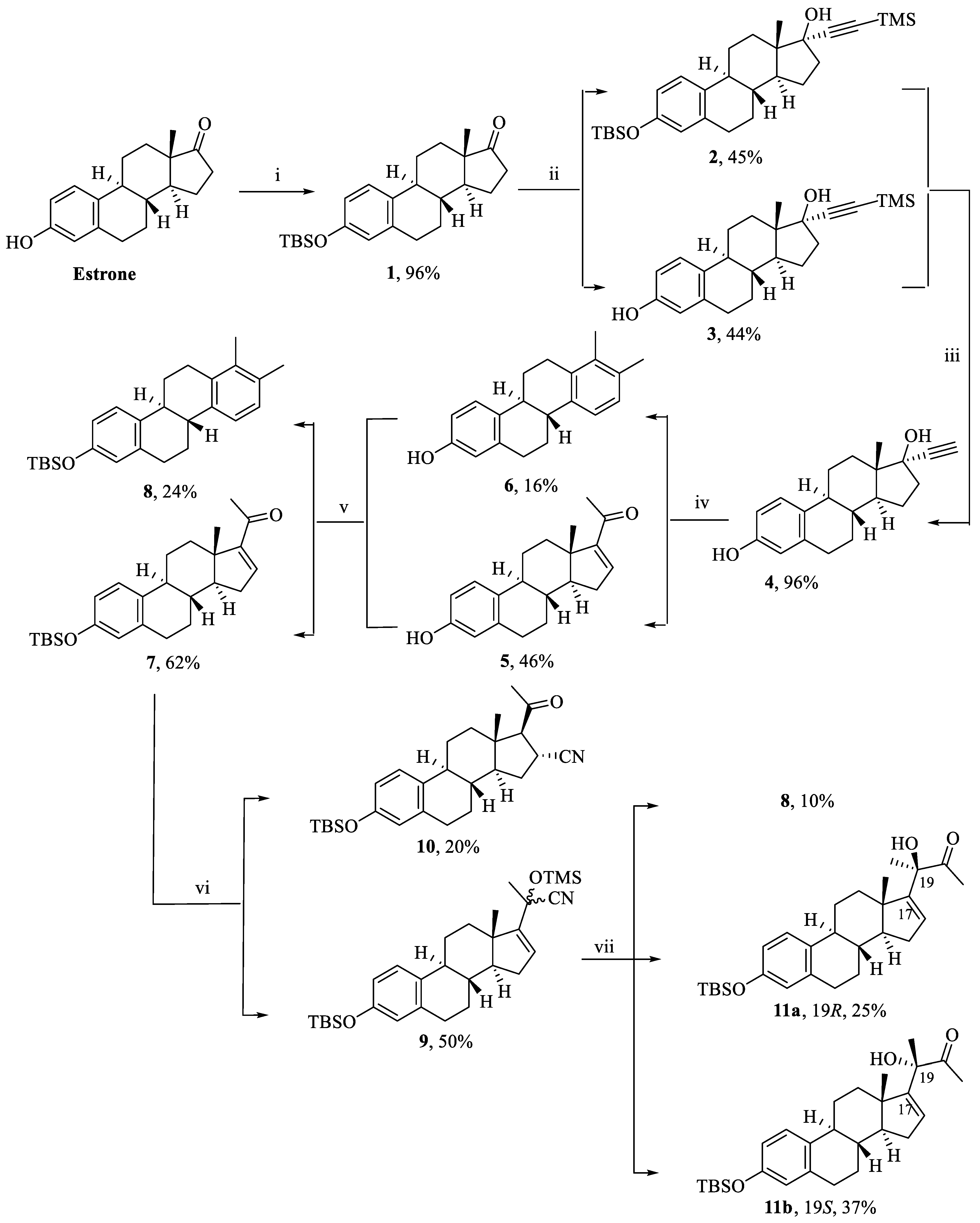
1-((8S,9S,13S,14S)-3-((tert-butyldimethylsilyl)oxy)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)ethenone (7). Following general procedure, A, compounds 6 and 5 (0.51 mmol) were protected with TBSCl (0.113 g, 0.75 mmol). White powder; 62% yield; 1H NMR (400 MHz, CDCl3) δ 6.92 (d, J = 8.4 Hz, 1H), 6.53 (dd, J = 3.3, 1.8 Hz, 1H), 6.42 (dd, J = 8.4, 2.5 Hz, 1H), 6.36 (d, J = 2.5 Hz, 1H), 2.72–2.52 (m, 2H), 2.35–2.30 (m, 1H), 2.22–2.12 (m, 2H), 2.08 (s, 3H), 1.96–1.89 (m, 1H), 1.73–1.67 (m, 1H), 1.46–1.07 (m, 6H), 0.79 (s, 9H), 0.73 (s, 3H), 0.00 (s, 6H).13C NMR (101 MHz, CDCl3) δ 196.72, 155.57, 153.35, 144.34, 137.58, 133.19, 125.94, 119.97, 117.21, 55.67, 46.53, 44.33, 36.97, 34.86, 32.03, 29.49, 27.84, 27.17, 26.43, 25.78, 18.21, 15.99, −4.32. 13C DEPT135-NMR (151 MHz, CDCl3) δ 144.31, 125.93, 119.95, 117.19, 55.68, 44.32, 36.97, 34.84 (inverted), 32.02 (inverted), 29.47 (inverted), 27.82 (inverted), 27.17, 26.40 (inverted), 25.74, 15.97, −4.36.
Tert-butyl(((4bS,10bR)-7,8-dimethyl-4b,5,6,10b,11,12-hexahydrochrysen-2-yl)oxy)dimethylsilane (8). Following general procedure, A, compound 8 was obtained as a side product. White powder; 24% yield; 1H NMR (600 MHz, CDCl3) δ 7.04 (d, J = 8.4 Hz, 1H), 7.00 (d, J = 8.0 Hz, 1H), 6.84 (d, J = 8.0 Hz, 1H), 6.47 (dd, J = 8.4, 2.6 Hz, 1H), 6.42 (d, J = 2.6 Hz, 1H), 2.83–2.70 (m, 3H), 2.69–2.61 (m, 1H), 2.52–2.38 (m, 4H), 2.09 (s, 3H), 1.98 (s, 3H), 1.50–1.42 (m, 2H), 0.79 (s, 9H), 0.00 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 211.42, 138.18, 137.99, 135.29, 134.77, 133.95, 133.03, 127.32, 126.42, 122.91, 119.83, 117.37, 41.54, 39.90, 30.20, 28.28, 27.90, 25.75, 20.45, 18.22, 15.12, 1.05, −4.34. 13C DEPT135-NMR (151 MHz, CDCl3) δ 127.32, 126.42, 122.91, 119.83, 117.37, 41.54, 39.90, 30.20 (inverted), 28.28 (inverted), 27.90 (inverted), 27.84 (inverted), 25.75, 20.46, −4.34.
2-((8S,9S,13S,14S)-3-((tert-butyldimethylsilyl)oxy)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-2-((trimethylsilyl)oxy)propanenitrile (9). Following general procedure, B, compound 7 was cyanosilylated. White powder; 50% yield; 1H NMR (400 MHz, CDCl3) δ 6.91 (d, J = 8.4 Hz, 1H), 6.42 (dd, J = 8.4, 2.5 Hz, 1H), 6.37 (d, J = 2.5 Hz, 1H), 5.74–5.70 (m, 1H), 2.68–2.60 (m, 2H), 2.17–1.97 (m, 3H), 1.84–1.67 (m, 2H), 1.58 (s, 3H), 1.51–1.00 (m, 6H), 0.84 (s, 3H), 0.79 (s, 9H), 0.08 (s, 9H), 0.00 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 152.97, 152.88, 151.95, 136.27, 131.68, 126.52, 126.30, 124.39, 120.36, 120.19, 118.57, 115.74, 70.99, 68.03, 67.26, 56.23, 56.03, 46.16, 45.65, 42.61, 42.35, 35.65, 34.18, 33.59, 30.54, 29.39, 29.22, 29.11, 28.99, 28.32, 28.05, 26.16, 25.00, 24.91, 24.32, 21.31, 16.76, 16.01, 15.94, 12.75, 0.17, 0.00, −0.37, −5.78.
(8S,9S,13S,14S)-17-acetyl-3-((tert-butyldimethylsilyl)oxy)-13-methyl-7,8,9,11,12,13,14,15,16,17-decahydro-6H-cyclopenta[a]phenanthrene-16-carbonitrile (10). Following general procedure, B, compound 10 was obtained as a side product. White powder; 20% yield; 1H NMR (400 MHz, CDCl3) δ 6.91 (d, J = 8.5 Hz, 1H), 6.44 (dd, J = 8.5, 2.4 Hz, 1H), 6.37 (d, J = 2.4 Hz, 1H), 3.41 (ddd, J = 11.6, 8.7, 2.9 Hz, 1H), 2.79 (d, J = 8.7 Hz, 1H), 2.65–2.60 (m, 2H), 2.21–2.08 (m, 2H), 2.04 (s, 3H), 2.00–1.94 (m, 1H), 1.65–1.53 (m, 3H), 1.37–1.21 (m, 3H), 0.79 (s, 9H), 0.42 (s, 3H), 0.00 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 204.72, 153.62, 137.47, 131.92, 126.02, 123.40, 120.09, 117.42, 69.03, 54.88, 45.31, 43.34, 38.52, 38.26, 31.11, 30.40, 29.36, 27.49, 26.27, 25.74, 25.38, 18.20, 13.43, −4.35. HRESI-MS m/z calcd for [M + H]+ C27H39NO2Si: 460.264227, found: 460.265381.
(R)-3-((8S,9S,13S,14S)-3-((tert-butyldimethylsilyl)oxy)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-3-hydroxybutan-2-one (11a). Following general procedure, C, R-isomer 11a was obtained. White powder; 25% yield; 1H NMR (400 MHz, CDCl3) δ 6.88 (d, J = 8.5 Hz, 1H), 6.41 (dd, J = 8.5, 2.5 Hz, 1H), 6.36 (d, J = 2.5 Hz, 1H), 5.71 (dd, J = 3.1, 1.3 Hz, 1H), 4.07 (s, 1H), 2.73–2.57 (m, 2H), 2.08−2.05 (m, 1H), 2.03 (s, 3H), 1.89–1.82 (m, 1H), 1.73–1.70 (m, 2H), 1.43–1.34 (m, 2H), 1.33 (s, 3H), 1.23–1.98 (m, 5H), 0.83 (s, 3H), 0.79 (s, 9H), 0.00 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 211.12, 155.63, 153.34, 137.70, 133.16, 128.82, 125.85, 119.98, 117.14, 79.55, 57.56, 47.81, 44.15, 37.06, 34.52, 31.08, 29.53, 27.68, 26.23, 25.77, 25.32, 23.32, 18.20, 17.28, −4.34.
(S)-3-((8S,9S,13S,14S)-3-((tert-butyldimethylsilyl)oxy)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-3-hydroxybutan-2-one (11b). Following general procedure, C, S-isomer 11b was obtained. White powder; 37% yield; 1H NMR (400 MHz, CDCl3) δ 6.90 (d, J = 8.6 Hz, 1H), 6.42 (dd, J = 8.4, 2.6 Hz, 1H), 6.36 (d, J = 2.4 Hz, 1H), 5.72–5.70 (m, 1H), 3.90 (s, 1H), 2.71–2.57 (m, 2H), 2.11–2.07 (m, 1H), 2.05 (s, 3H), 1.82–1.76 (m, 2H), 1.72–1.65 (m, 2H), 1.55–1.48 (m, 2H), 1.37 (s, 3H), 1.28–1.03 (m, 4H), 0.79 (s, 9H), 0.59 (s, 3H), 0.00 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 210.99, 155.56, 153.32, 137.69, 133.22, 128.78, 125.86, 119.97, 117.15, 80.09, 56.73, 48.14, 43.93, 37.15, 36.20, 31.06, 29.49, 27.67, 26.46, 25.75, 24.92, 23.74, 18.20, 17.00, −4.35.
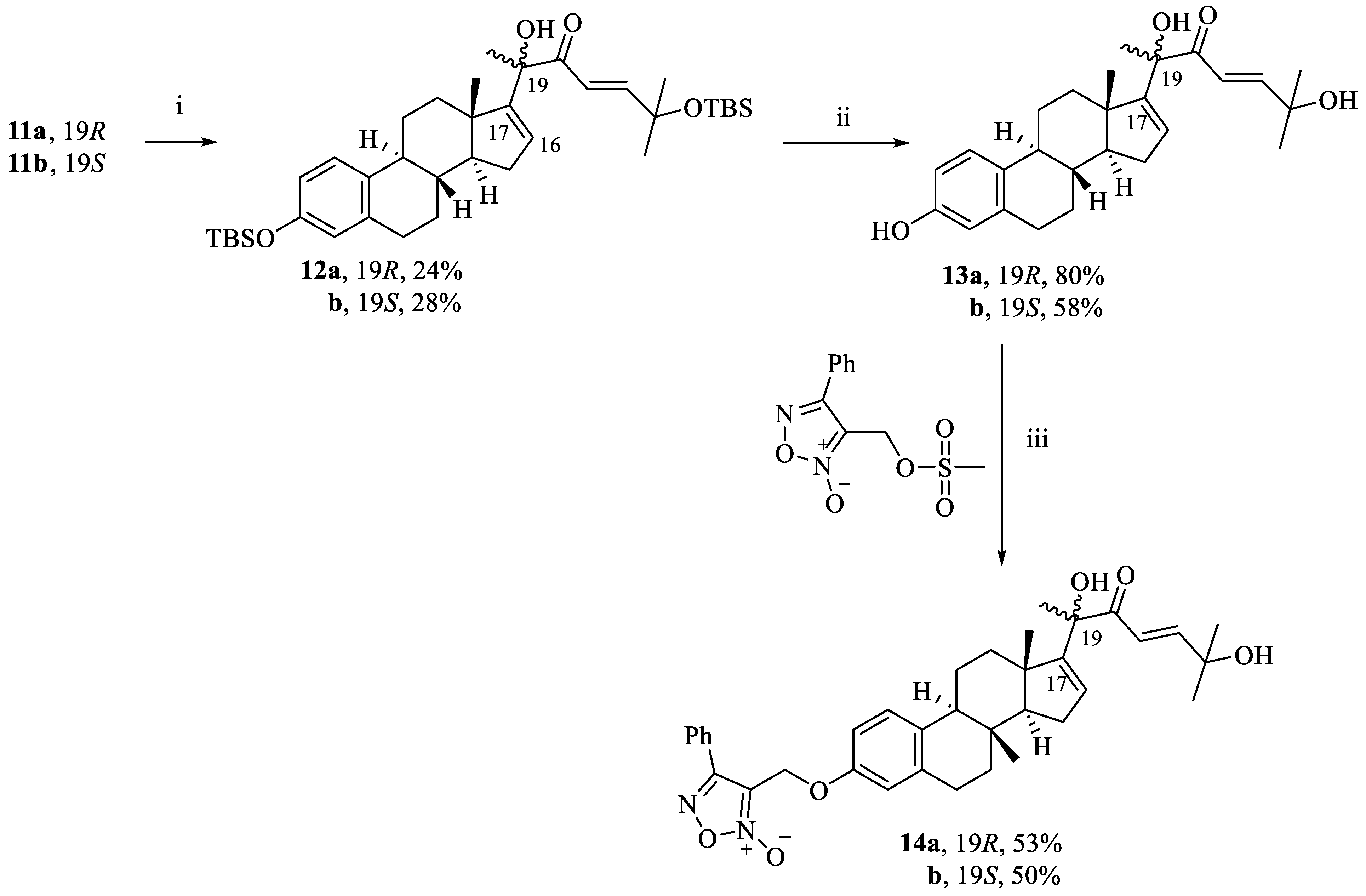
(R,E)-6-((tert-butyldimethylsilyl)oxy)-2-((8S,9S,13S,14S)-3-((tert-butyldimethylsilyl)oxy)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-2-hydroxy-6-methylhept-4-en-3-one (12a). Following general procedure, D, compound 11a (0.500 g, 0.79 mmol) was used as a starting material. White powder; 24% yield; 1H NMR (600 MHz, CDCl3) δ 6.88 (dd, J = 15.1, 8.4 Hz, 2H), 6.55 (d, J = 15.1 Hz, 1H), 6.41 (dd, J = 8.4, 2.4 Hz, 1H), 6.37 (d, J = 2.4 Hz, 1H), 5.73 (d, J = 1.9 Hz, 1H), 4.21 (s, 1H), 2.72–2.58 (m, 2H), 2.06–1.97 (m, 2H), 1.94–1.85 (m, 2H), 1.74–1.62 (m, 2H), 1.44–1.32 (m, 3H), 1.30 (s, 3H), 1.17, 1.16 (two s, 6H), 1.11–0.97 (m, 2H), 0.84 (s, 3H), 0.79 (s, 9H), 0.73 (s, 9H), 0.00 (s, 6H), −0.08 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 203.99, 158.57, 157.14, 155.36, 139.78, 135.22, 131.33, 127.85, 122.03, 120.90, 119.15, 80.53, 75.64, 59.60, 49.57, 46.30, 39.02, 36.17, 33.07, 31.90, 31.53, 29.69, 28.18, 27.87, 27.78, 27.25, 20.23, 19.35, 0.00, −0.04, −2.33.
(S,E)-6-((tert-butyldimethylsilyl)oxy)-2-((8S,9S,13S,14S)-3-((tert-butyldimethylsilyl)oxy)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-2-hydroxy-6-methylhept-4-en-3-one (12b). Following general procedure, D, compound 11b (0.500 g, 0.79 mmol) was used as a starting material. White powder; 28% yield; 1H NMR (600 MHz, CDCl3) δ 6.90 (d, J = 8.5 Hz, 1H), 6.85 (d, J = 15.1 Hz, 1H), 6.53 (d, J = 15.1 Hz, 1H), 6.42 (dd, J = 8.5, 2.5 Hz, 1H), 6.36 (d, J = 2.5 Hz, 1H), 5.73 (d, J = 1.8 Hz, 1H), 4.12 (s, 1H), 2.71–2.57 (m, 2H), 2.11–2.00 (m, 3H), 1.80–1.66 (m, 4H), 1.55–1.50 (m, 1H), 1.34 (s, 3H), 1.30–1.19 (m, 2H), 1.17 (s, 3H), 1.14 (s, 3H), 1.08–1.06 (m, 1H), 0.79 (s, 9H), 0.73 (s, 9H), 0.53 (s, 3H), 0.00 (s, 6H), −0.07, −0.09 (two s, 6H). 13C NMR (151 MHz, CDCl3) δ 203.67, 157.94, 157.51, 155.36, 139.80, 135.41, 131.26, 127.95, 122.03, 121.46, 119.19, 81.27, 75.63, 58.90, 50.15, 46.05, 39.24, 38.44, 33.10, 31.94, 31.75, 31.59, 29.76, 28.55, 27.90, 27.83, 27.14, 20.30, 20.28, 19.30, 0.00, −0.02, −2.29.
(R,E)-2,6-dihydroxy-2-((8S,9S,13S,14S)-3-hydroxy-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-6-methylhept-4-en-3-one (13a). Following general procedure, F, compound 12a (0.636 g, 1 mmol) was deprotected using TBAF (3.1 mL, 3.1 mmol). White powder; 80% yield; 1H NMR (600 MHz, Acetone) δ 7.81 (s, 1H), 6.97 (d, J = 15.4 Hz, 1H), 6.90 (d, J = 8.4 Hz, 1H), 6.65 (d, J = 15.5 Hz, 1H), 6.44 (dd, J = 8.4, 2.6 Hz, 1H), 6.38 (d, J = 2.6 Hz, 1H), 5.83 (dd, J = 3.3, 1.5 Hz, 1H), 4.33 (s, 1H), 3.97 (s, 1H), 2.71–2.59 (m, 2H), 2.12–2.04 (m, 2H), 1.97–1.87 (m, 3H), 1.77–1.71 (m, 2H), 1.44–1.39 (m, 1H), 1.32 (s, 3H), 1.24–1.20 (m, 1H), 1.19 (s, 3H), 1.18 (s, 3H), 1.16–1.12 (m, 2H), 0.87 (s, 3H). 13C NMR (151 MHz, Acetone) δ 202.03, 156.66, 156.60, 155.98, 138.35, 132.09, 129.19, 126.76, 120.04, 116.01, 113.58, 79.41, 70.84, 58.17, 48.44, 45.04, 38.28, 35.15, 31.65, 30.27, 29.50, 28.48, 27.16, 25.89, 17.65.
(S,E)-2,6-dihydroxy-2-((8S,9S,13S,14S)-3-hydroxy-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-6-methylhept-4-en-3-one (13b). Following general procedure, F, compound 12b (0.636 g, 1 mmol) was deprotected using TBAF (3.1 mL, 3.1 mmol). White powder; 58% yield; 1H NMR (600 MHz, Acetone) δ 7.80 (s, 1H), 6.92 (dd, J = 15.6, 8.4 Hz, 2H), 6.69 (d, J = 15.6 Hz, 1H), 6.45 (dd, J = 8.4, 2.6 Hz, 1H), 6.39 (d, J = 2.6 Hz, 1H), 5.73 (dd, J = 3.2, 1.4 Hz, 1H), 4.26 (s, 1H, C-19-OH), 3.92 (s, 1H), 2.70–2.59 (m, 2H), 2.16–2.04 (m, 2H), 1.93–1.82 (m, 3H), 1.78–1.73 (m, 1H), 1.63–1.58 (m, 1H), 1.51–1.46 (m, 1H), 1.43–1.37 (m, 1H), 1.35 (s, 3H), 1.31–1.21 (m, 2H), 1.18 (s, 3H), 1.17 (s, 3H), 0.69 (s, 3H). 13C NMR (151 MHz, Acetone) δ 201.24, 157.05, 155.98, 155.48, 138.35, 132.18, 128.38, 126.76, 120.89, 116.00, 113.57, 79.99, 70.76, 57.93, 48.72, 44.96, 38.35, 36.94, 31.58, 30.26, 29.49, 28.51, 27.35, 25.71, 17.65.
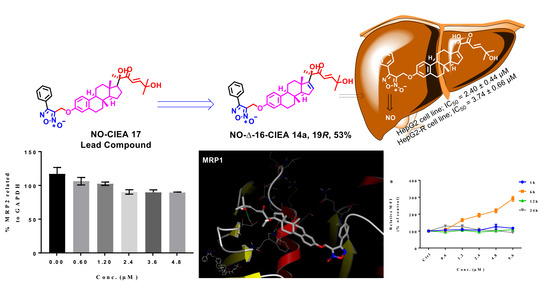
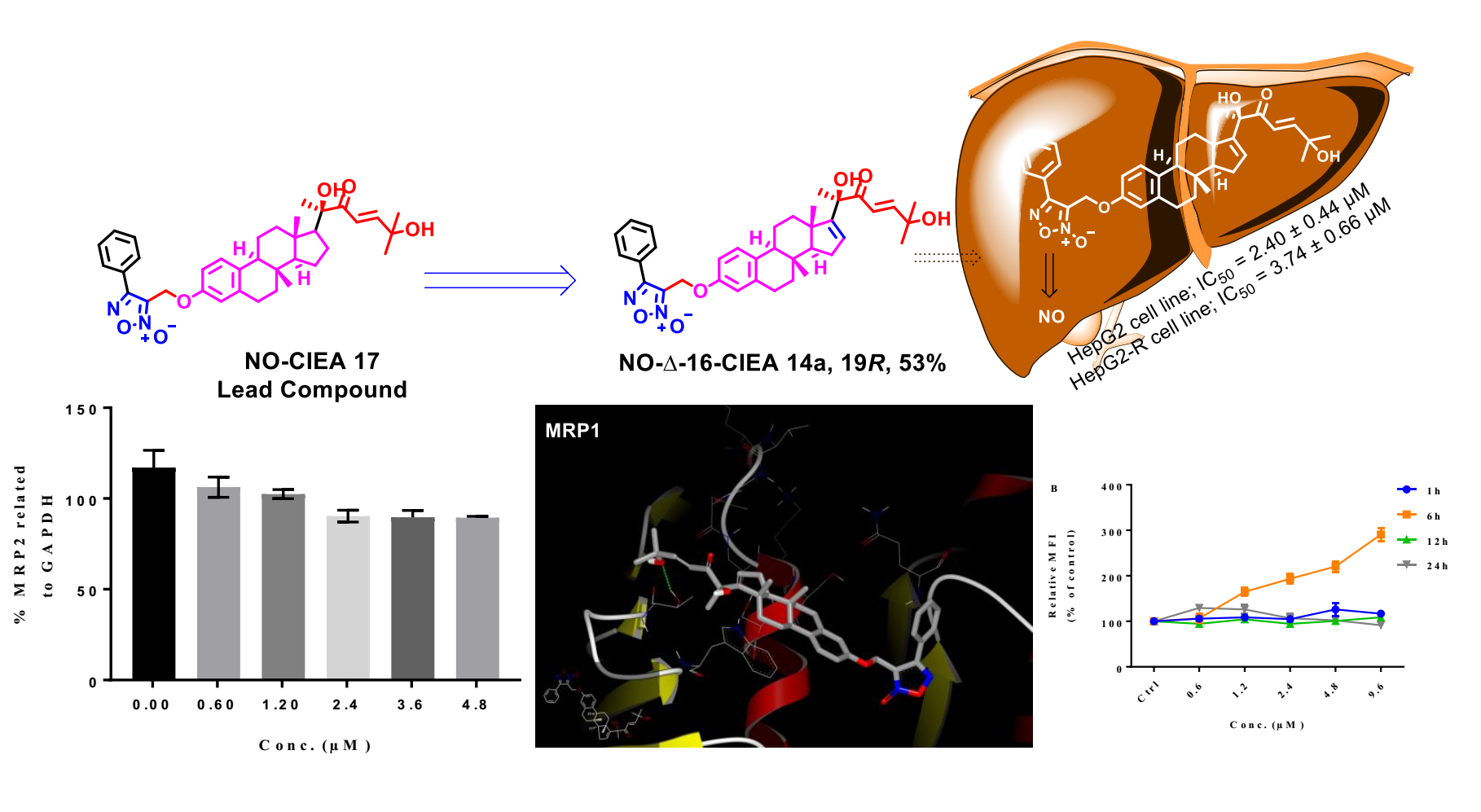
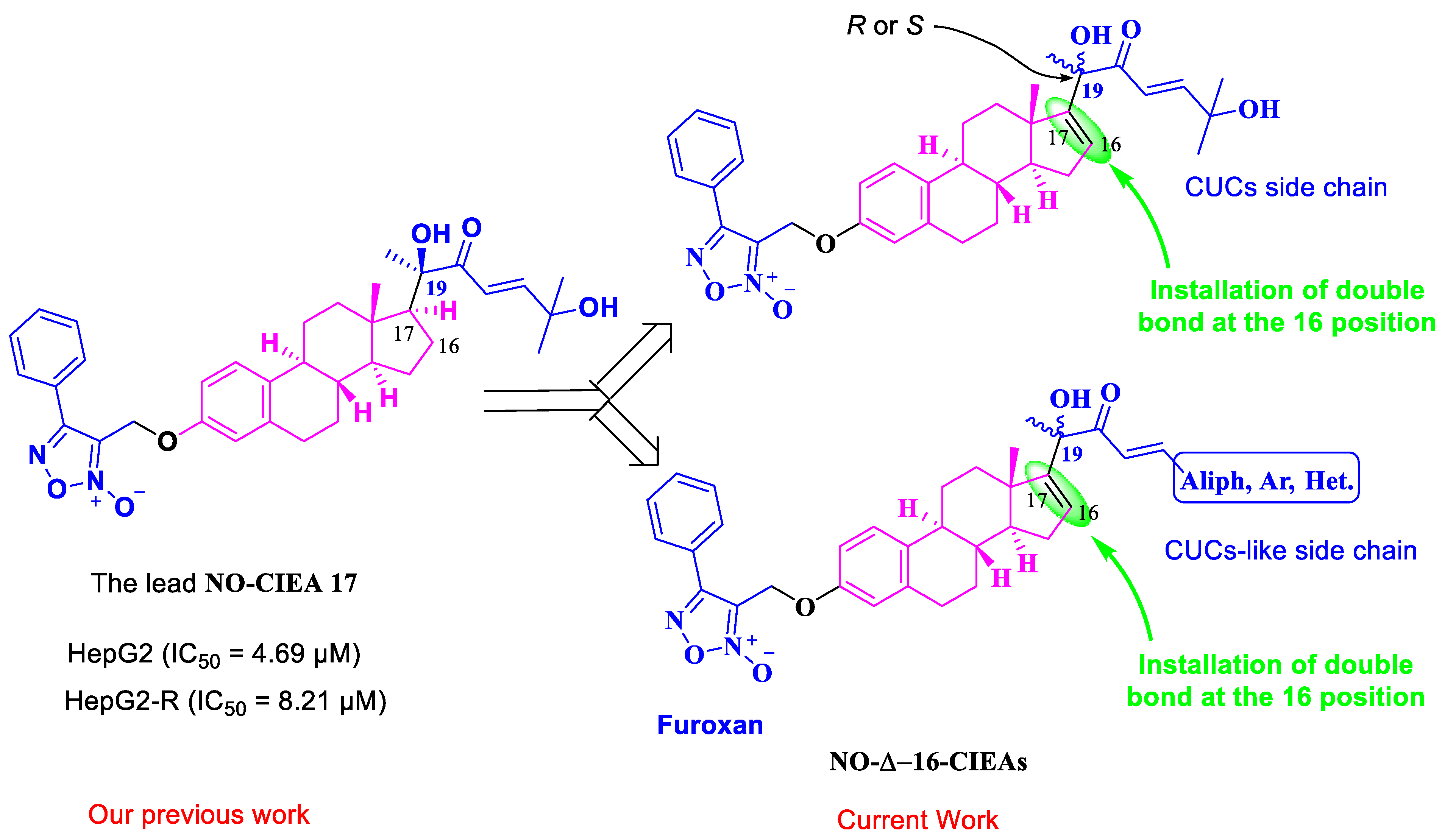
3-((((8S,9S,13S,14S)-17-((R,E)-2,6-dihydroxy-6-methyl-3-oxohept-4-en-2-yl)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-3-yl)oxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (14a). Following general procedure, G, compound 13a (0.115 g, 0.28 mmol) was alkylated with furoxan mesylate. Yellowish green powder; 53% yield; 1H NMR (600 MHz, CDCl3) δ 7.78 (d, J = 7.0 Hz, 2H), 7.50–7.42 (m, 3H), 7.10–7.06 (two d, J = 8.6, 15.4 Hz, 2H), 6.70 (dd, J = 8.6, 2.7 Hz, 1H), 6.64 (d, J = 2.7 Hz, 1H), 6.58 (d, J = 15.4 Hz, 1H), 5.87 (d, J = 1.8 Hz, 1H), 4.98 (s, 2H), 4.31 (s, 1H), 2.84–2.72 (m, 2H), 2.20–1.95 (m, 4H), 1.86–1.77 (m, 2H), 1.55–1.49 (m, 2H), 1.43 (s, 3H), 1.32 (s, 3H), 1.31 (s, 3H), 1.24–1.07 (m, 3H), 0.94 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 201.54, 157.16, 155.30, 154.85, 154.84, 138.48, 134.64, 131.36, 129.48, 129.35, 128.62, 127.77, 126.42, 126.25, 118.77, 114.93, 112.25, 78.63, 71.32, 58.30, 57.39, 47.70, 44.10, 36.94, 34.28, 31.10, 29.64, 29.50, 29.46, 27.46, 26.18, 25.20, 17.25. 13C DEPT135-NMR (151 MHz, CDCl3) δ 155.31, 131.37, 129.49, 129.36, 127.77, 126.43, 118.76, 114.93, 112.25, 58.29 (inverted), 57.38, 44.10, 36.94, 34.28 (inverted), 31.10 (inverted), 29.64 (inverted), 29.51, 27.46 (inverted), 26.18 (inverted), 25.19, 17.24. HRESI-MS m/z calcd for [M + Na]+ C35H40N2O6: 607.277858, found: 607.279321.
3-((((8S,9S,13S,14S)-17-((S,E)-2,6-dihydroxy-6-methyl-3-oxohept-4-en-2-yl)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-3-yl)oxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (14b). Following general procedure, G, compound 13b (0.115 g, 0.28 mmol) was alkylated with furoxan mesylate. Yellowish green powder; 50% yield; 1H NMR (600 MHz, CDCl3) δ 7.82–7.76 (m, 2H), 7.50–7.41 (m, 3H), 7.12 (d, J = 8.6 Hz, 1H), 7.05 (d, J = 15.4 Hz, 1H), 6.70 (dd, J = 8.6, 2.7 Hz, 1H), 6.64 (d, J = 2.7 Hz, 1H), 6.58 (d, J = 15.4 Hz, 1H), 5.86 (dd, J = 3.1, 1.3 Hz, 1H), 4.99 (s, 2H), 4.17 (s, 1H), 2.84–2.74 (m, 2H), 2.20–2.16 (m, 2H), 1.94–1.86 (m, 2H), 1.84–1.77 (m, 2H), 1.65–1.51 (m, 2H), 1.47 (s, 3H), 1.36–1.33 (m, 1H), 1.31 (s, 3H), 1.28 (s, 3H), 1.24–1.18 (m, 2H), 0.64 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 201.30, 157.16, 155.24, 154.84, 154.73, 138.45, 134.74, 131.36, 129.35, 129.19, 127.77, 126.47, 126.25, 119.37, 114.90, 112.26, 112.24, 79.17, 71.24, 58.30, 56.74, 48.06, 43.90, 37.08, 36.19, 31.08, 29.61, 29.47, 29.34, 27.48, 26.46, 24.93, 17.34. 13C DEPT135-NMR (151 MHz, CDCl3) δ 154.74, 131.36, 129.35, 129.20, 127.77, 126.48, 119.36, 114.89, 112.25, 58.29 (inverted), 56.74, 43.89, 37.08, 36.19 (inverted), 31.08 (inverted), 29.62 (inverted), 29.34, 27.48 (inverted), 26.46 (inverted), 24.92, 17.34. HRESI-MS m/z calcd for [M + Na]+ C35H40N2O6: 607.277858, found: 607.279500.
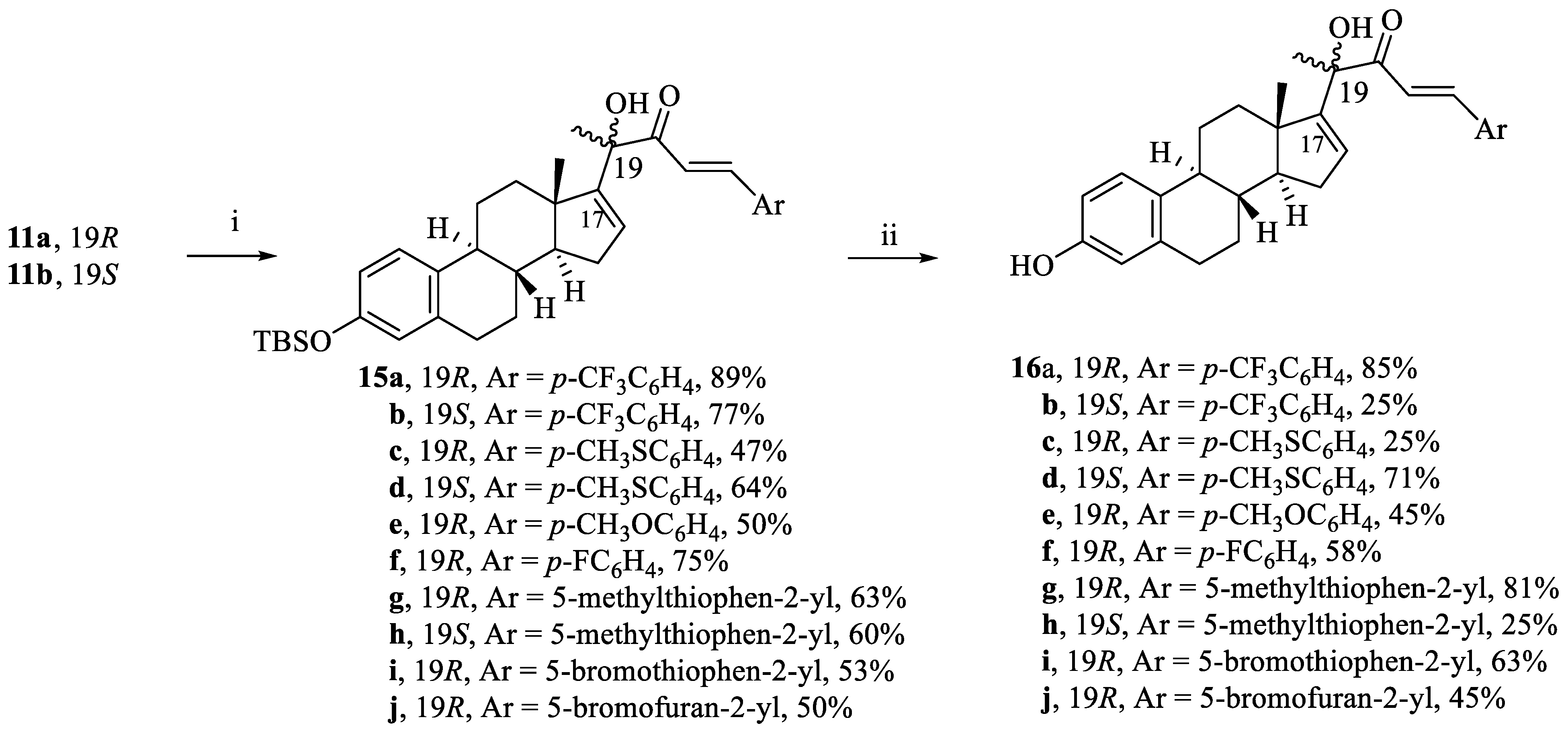
(R,E)-4-((8S,9S,13S,14S)-3-((tert-butyldimethylsilyl)oxy)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-4-hydroxy-1-(4-(trifluoromethyl)phenyl)pent-1-en-3-one (15a). Following general procedure, E, compound 11a (0.455 g, 1 mmol) underwent aldol condensation using p-trifluoromethylbenzaldehyde (0.261 g, 1.5 mmol). White powder; 89% yield; 1H NMR (400 MHz, CDCl3) δ 7.68 (d, J = 15.9 Hz, 1H), 7.57–7.45 (m, 4H), 6.92 (d, J = 15.9 Hz, 1H), 6.86 (d, J = 8.4 Hz, 1H), 6.41 (dd, J = 8.4, 2.5 Hz, 1H), 6.36 (d, J = 2.5 Hz, 1H), 5.86 (d, J = 1.7 Hz, 1H), 4.23 (s, 1H), 2.73–2.56 (m, 2H), 2.16–2.09 (m, 1H), 2.04–1.88 (m, 2H), 1.74–1.71 (m, 2H), 1.48–1.43 (m, 1H), 1.41 (s, 3H), 1.38–0.97 (m, 5H), 0.88 (s, 3H), 0.79 (s, 9H), 0.00 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 200.70, 155.16, 153.29, 142.93, 137.70, 133.15, 132.12, 129.75, 128.80, 125.99, 125.81, 122.40, 121.10, 119.94, 117.09, 78.79 (C-19-OH), 57.53, 47.85, 43.98, 37.03, 34.50, 31.24, 29.50, 27.60, 26.19, 25.72, 25.25, 18.18, 17.25, −4.38.
(S,E)-4-((8S,9S,13S,14S)-3-((tert-butyldimethylsilyl)oxy)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-4-hydroxy-1-(4-(trifluoromethyl)phenyl)pent-1-en-3-one (15b). Following general procedure, E, compound 11b (0.455 g, 1 mmol) underwent aldol condensation using p-trifluoromethylbenzaldehyde (0.261 g, 1.5 mmol). White powder; 77% yield; 1H NMR (400 MHz, CDCl3) δ 7.64 (d, J = 15.8 Hz, 1H), 7.53–7.43 (m, 4H), 6.91 (dd, J = 15.8, 8.4 Hz, 2H), 6.42 (dd, J = 8.4, 2.5 Hz, 1H), 6.37 (d, J = 2.5 Hz, 1H), 5.82 (d, J = 1.8 Hz, 1H), 4.01 (s, 1H), 2.69–2.63 (m, 2H), 2.14–2.03 (m, 3H), 1.85–1.67 (m, 2H), 1.60–1.49 (m, 1H), 1.43 (s, 3H), 1.40–1.03 (m, 5H), 0.79 (s, 9H), 0.59 (s, 3H), 0.00 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 200.40, 155.57, 153.35, 142.32, 137.68, 133.17, 132.08, 129.31, 128.69, 125.96, 125.92, 125.86, 121.87, 119.97, 117.15, 79.38, 56.92, 48.16, 43.96, 37.19, 31.98, 29.76, 29.72, 29.42, 25.73, 22.74, 18.18, 17.54, 14.16, −4.38.
(R,E)-4-((8S,9S,13S,14S)-3-((tert-butyldimethylsilyl)oxy)-13-methyl 7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-4-hydroxy-1-(4-(methylthio)phenyl)pent-1-en-3-one (15c). Following general procedure, E, compound 11a (0.455 g, 1 mmol) underwent aldol condensation using 4-(methylthio)benzaldehyde (0.228 g, 1.5 mmol). Yellow powder; 47% yield; 1H NMR (400 MHz, CDCl3) δ 7.63 (d, J = 15.8 Hz, 1H), 7.32 (d, J = 8.4 Hz, 2H), 7.06 (d, J = 8.4 Hz, 2H), 6.86 (d, J = 8.4 Hz, 1H), 6.80 (d, J = 15.8 Hz, 1H), 6.40 (dd, J = 8.4, 2.5 Hz, 1H), 6.36 (d, J = 2.5 Hz, 1H), 5.83 (s, J = 1.7 Hz, 1H), 4.34 (s, 1H), 2.73–2.56 (m, 2H), 2.32 (s, 3H), 2.14–2.08 (m, 1H), 2.03–1.86 (m, 2H), 1.75–1.68 (m, 2H), 1.46–1.44 (m, 1H), 1.39 (s, 3H), 1.35–1.09 (m, 5H) 0.87 (s, 3H), 0.79 (s, 9H), 0.00 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 200.85, 155.52, 153.25, 144.46, 143.16, 137.72, 133.29, 130.74, 129.24, 129.09, 125.90, 125.84, 119.92, 117.70, 117.07, 78.56, 57.44, 47.81, 43.96, 37.05, 34.38, 31.21, 29.53, 27.62, 26.22, 25.75, 25.47, 18.19, 17.28, 15.08, −4.35.
(S,E)-4-((8S,9S,13S,14S)-3-((tert-butyldimethylsilyl)oxy)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-4-hydroxy-1-(4-(methylthio)phenyl)pent-1-en-3-one (15d). Following general procedure, E, compound 11b (0.455 g, 1 mmol) underwent aldol condensation using 4-(methylthio)benzaldehyde (0.228 g, 1.5 mmol). Yellow powder; 64% yield; 1H NMR (600 MHz, CDCl3) δ 7.60 (d, J = 15.7 Hz, 1H), 7.30 (d, J = 8.4 Hz, 2H), 7.05 (d, J = 8.4 Hz, 2H), 6.91 (d, J = 8.5 Hz, 1H), 6.79 (d, J = 15.7 Hz, 1H), 6.42 (dd, J = 8.5, 2.6 Hz, 1H), 6.36 (d, J = 2.6 Hz, 1H), 5.80 (dd, J = 3.2, 1.4 Hz, 1H), 4.17 (s, 1H), 2.69–2.60 (m, 2H), 2.32 (s, 3H), 2.12–2.04 (m, 3H), 1.87–1.79 (m, 2H), 1.71–1.69 (m, 2H), 1.57–1.52 (m, 1H), 1.42 (s, 3H), 1.31–1.20 (m, 2H), 1.08 (s, 1H), 0.79 (s, 9H), 0.57 (s, 3H), 0.00 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 200.66, 155.87, 153.31, 143.90, 143.03, 137.72, 133.29, 130.79, 129.00, 128.94, 125.93, 125.87, 119.95, 118.46, 117.12, 79.13, 56.92, 48.15, 43.96, 37.17, 36.28, 31.15, 29.50, 27.67, 26.47, 25.75, 25.18, 18.20, 17.51, 15.10, −4.35.
(R,E)-4-((8S,9S,13S,14S)-3-((tert-butyldimethylsilyl)oxy)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-1-(4-methoxyphenyl)-4-hydroxypent-1-en-3-one (15e). Following general procedure, E, compound 11a (0.455 g, 1 mmol) underwent aldol condensation using 4-methoxybenzaldehyde (0.204 g, 1.5 mmol). White powder; 50% yield; 1H NMR (600 MHz, CDCl3) δ 7.65 (d, J = 15.7 Hz, 1H), 7.38 (d, J = 8.8 Hz, 2H), 6.86 (d, J = 8.5 Hz, 1H), 6.76 (d, J = 8.8 Hz, 2H), 6.73 (d, J = 15.7 Hz, 1H), 6.40 (dd, J = 8.5, 2.5 Hz, 1H), 6.36 (d, J = 2.5 Hz, 1H), 5.82 (d, J = 1.8 Hz, 1H), 4.38 (s, 1H), 3.68 (s, 3H), 2.71–2.56 (m, 2H), 2.12–2.08 (m, 1H), 2.02–1.85 (m, 3H), 1.74–1.66 (m, 2H), 1.45–1.41 (m, 1H), 1.38 (s, 3H), 1.37–1.31 (m, 2H), 1.23–1.16 (m, 1H), 1.10–1.02 (m, 1H), 0.88 (s, 3H), 0.79 (s, 9H), 0.00 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 200.87, 162.04, 155.65, 153.24, 144.77, 137.73, 133.32, 130.58, 129.05, 127.08, 125.84, 119.90, 117.05, 116.46, 114.49, 78.46, 57.40, 55.47, 47.80, 43.95, 37.06, 34.33, 31.18, 29.53, 27.61, 26.21, 25.73, 25.53, 18.18, 17.26, −4.37.
(R,E)-4-((8S,9S,13S,14S)-3-((tert-butyldimethylsilyl)oxy)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-1-(4-fluorophenyl)-4-hydroxypent-1-en-3-one (15f). Following general procedure, E, compound 11a (0.455 g, 1 mmol) underwent aldol condensation using 4-fluorobenzaldehyde (0.186 g, 1.5 mmol). White powder; 75% yield; 1H NMR (600 MHz, CDCl3) δ 7.64 (d, J = 15.8 Hz, 1H), 7.43–7.38 (m, 2H), 6.96–6.91 (m, 2H), 6.86 (d, J = 8.4 Hz, 1H), 6.78 (d, J = 15.8 Hz, 1H), 6.40 (dd, J = 8.4, 2.6 Hz, 1H), 6.36 (d, J = 2.6 Hz, 1H), 5.83 (dd, J = 3.2, 1.4 Hz, 1H), 4.29 (s, 1H), 2.70–2.58 (m, 2H), 2.13–2.09 (m, 1H), 2.02–1.88 (m, 3H), 1.73–1.69 (m, 2H), 1.45–1.41 (m, 1H), 1.39 (s, 3H), 1.36–1.34 (m, 1H), 1.23 –1.04 (m, 3H), 0.88 (s, 3H), 0.79 (s, 9H), −0.00 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 200.80, 155.41, 153.27, 143.65, 137.70, 133.22, 130.72, 130.66, 129.37, 125.80, 119.91, 118.52, 117.07, 116.30, 116.16, 78.61, 57.48, 47.83, 43.98, 37.04, 34.41, 31.20, 29.50, 27.61, 26.19, 25.71, 25.37, 18.17, 17.25, −4.38.
(R,E)-4-((8S,9S,13S,14S)-3-((tert-butyldimethylsilyl)oxy)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-4-hydroxy-1-(5-methylthiophen-2-yl)pent-1-en-3-one (15g). Following general procedure, E, compound 11a (0.455 g, 1 mmol) underwent aldol condensation using 5-methylthiophene-2-carbaldehyde (0.189 g, 1.5 mmol). Yellow powder; 63% yield; 1H NMR (600 MHz, CDCl3) δ 7.69 (d, J = 15.4 Hz, 1H), 6.99 (d, J = 3.6 Hz, 1H), 6.86 (d, J = 8.5 Hz, 1H), 6.57 (dd, J = 3.6, 1.1 Hz, 1H,), 6.48 (d, J = 15.4 Hz, 1H), 6.40 (dd, J = 8.5, 2.5 Hz, 1H), 6.36 (d, J = 2.5 Hz, 1H), 5.80 (dd, J = 3.2, 1.4 Hz, 1H), 4.33 (s, 1H), 2.71–2.56 (m, 2H), 2.34 (s, 3H), 2.12–2.08 (m, 1H), 2.01–1.86 (m, 3H), 1.73–1.66 (m, 2H), 1.43–1.38 (m, 1H), 1.36 (s, 3H), 1.24–1.02 (m, 4H), 0.86 (s, 3H), 0.79 (s, 9H), −0.00 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 200.63, 155.50, 153.24, 145.39, 137.90, 137.74, 137.66, 133.59, 133.33, 129.12, 127.03, 125.84, 119.90, 117.05, 116.41, 78.37, 57.39, 47.77, 43.97, 37.06, 34.32, 31.17, 29.54, 27.62, 26.21, 25.73, 25.53, 18.18, 17.25, 15.97, −4.37.
(S,E)-4-((8S,9S,13S,14S)-3-((tert-butyldimethylsilyl)oxy)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-4-hydroxy-1-(5-methylthiophen-2-yl)pent-1-en-3-one (15h). Following general procedure, E, compound 11b (0.455 g, 1 mmol) underwent aldol condensation using 5-methylthiophene-2-carbaldehyde (0.189 g, 1.5 mmol). Yellow powder; 60% yield; 1H NMR (400 MHz, CDCl3) δ 7.66 (d, J = 15.4 Hz, 1H), 6.97 (d, J = 3.6 Hz, 1H), 6.91 (d, J = 8.5 Hz, 1H), 6.56 (dd, J = 3.6, 1.1 Hz, 1H), 6.47 (d, J = 15.4 Hz, 1H), 6.42 (dd, J = 8.5, 2.4 Hz, 1H), 6.36 (d, J = 2.4 Hz, 1H), 5.83–5.75 (m, 1H), 4.19 (s, 1H), 2.70–2.58 (m, 2H), 2.33 (s, 3H), 2.13–2.00 (m, 3H), 1.87–1.78 (m, 2H), 1.75–1.67 (m, 2H), 1.57–1.50 (m, 1H), 1.39 (s, 3H), 1.30–1.03 (m, 3H), 0.79 (s, 9H), 0.57 (s, 3H), 0.00 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 200.42, 155.85, 153.29, 145.26, 137.90, 137.74, 137.20, 133.52, 133.32, 128.88, 127.01, 125.87, 119.94, 117.13, 117.10, 78.96, 56.90, 48.14, 43.96, 37.16, 36.30, 31.13, 29.51, 27.67, 26.46, 25.74, 25.25, 18.19, 17.46, 15.96, −4.36.
(R,E)-1-(5-bromothiophen-2-yl)-4-((8S,9S,13S,14S)-3-((tert-butyldimethylsilyl)oxy)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-4-hydroxypent-1-en-3-one (15i). Following general procedure, E, compound 11a (0.455 g, 1 mmol) underwent aldol condensation using 5-bromothiophene-2-carbaldehyde (0.286 g, 1.5 mmol). Yellow powder; 53% yield; 1H NMR (600 MHz, CDCl3) δ 7.63 (d, J = 15.4 Hz, 1H), 6.92 (d, J = 3.9 Hz, 1H), 6.87–6.85 (m, 2H), 6.51 (d, J = 15.4 Hz, 1H), 6.40 (dd, J = 8.5, 2.6 Hz, 1H), 6.36 (d, J = 2.6 Hz, 1H), 5.80 (dd, J = 3.2, 1.3 Hz, 1H), 4.23 (s, 1H), 2.72–2.55 (m, 2H), 2.12- 2.08 (m, 1H), 2.02–1.85 (m, 3H), 1.74–1.65 (m, 2H), 1.44–1.38 (m, 1H), 1.36 (s, 3H), 1.34–1.32 (m, 1H), 1.20–1.01 (m, 3H), 0.86 (s, 3H), 0.79 (s, 9H), 0.00 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 200.46, 155.24, 153.27, 141.38, 137.74, 136.24, 133.25, 132.97, 131.45, 129.50, 125.82, 119.93, 117.98, 117.15, 117.07, 78.53, 57.47, 47.80, 43.98, 37.05, 34.42, 31.21, 29.52, 27.60, 26.21, 25.74, 25.39, 18.19, 17.24, −4.36.
(R,E)-1-(5-bromofuran-2-yl)-4-((8S,9S,13S,14S)-3-((tert-butyldimethylsilyl)oxy)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-4-hydroxypent-1-en-3-one (15j). Following general procedure, E, compound 11a (0.455 g, 1 mmol) underwent aldol condensation using 5-bromofuran-2-carbaldehyde (0.261 g, 1.5 mmol). Yellow powder; 50% yield; 1H NMR (600 MHz, CDCl3) δ 7.29 (d, J = 15.4 Hz, 1H), 6.86 (d, J = 8.5 Hz, 1H), 6.64 (d, J = 15.4 Hz, 1H), 6.49 (d, J = 3.5 Hz, 1H), 6.40 (dd, J = 8.5, 2.5 Hz, 1H), 6.36 (d, J = 2.5 Hz, 1H), 6.27 (d, J = 3.5 Hz, 1H), 5.83 (dd, J = 3.1, 1.3 Hz, 1H), 4.28 (s, 1H), 2.71–2.57 (m, 2H), 2.17–2.09 (m, 1H), 2.03–1.85 (m, 3H), 1.74–1.66 (m, 2H), 1.45–1.38 (m, 1H), 1.37 (s, 3H), 1.35–1.30 (m, 1H), 1.22–0.96 (m, 3H), 0.86 (s, 3H), 0.79 (s, 9H), −0.00 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 200.69, 154.96, 153.25, 153.05, 137.78, 133.32, 129.60, 129.41, 126.42, 125.81, 119.92, 119.17, 117.05, 116.74, 114.81, 78.60, 57.21, 47.79, 43.93, 37.05, 34.45, 31.18, 29.54, 27.60, 26.23, 25.73, 25.35, 18.18, 17.26, −4.37.
(R,E)-4-hydroxy-4-((8S,9S,13S,14S)-3-hydroxy-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-1-(4-(trifluoromethyl)phenyl)pent-1-en-3-one (16a). Following general procedure, F, compound 15a (0.305 g, 0.5 mmol) was deprotected using TBAF (1.55 mL, 1.55 mmol). White powder; 63% yield; 1H NMR (600 MHz, CDCl3) δ 7.77 (d, J = 15.8 Hz, 1H), 7.64–7.53 (m, 4H), 7.01 (d, J = 15.8 Hz, 1H), 6.97 (d, J = 8.4 Hz, 1H), 6.52 (dd, J = 8.4, 2.5 Hz, 1H), 6.47 (d, J = 2.5 Hz, 1H), 5.96 (d, J = 1.8 Hz, 1H), 5.04 (s, 1H), 4.38 (s, 1H), 2.80–2.67 (m, 2H), 2.24–2.19 (m, 1H), 2.10–1.98 (m, 3H), 1.82–1.79 (m, 2H), 1.51 (s, 3H), 1.47–1.40 (m, 2H), 1.32–1.06 (m, 3H), 0.97 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 200.73, 155.03, 153.41, 143.11, 138.11, 137.63, 132.70, 129.88, 128.83, 126.19, 125.99, 125.97, 121.01, 115.28, 112.66, 78.87, 57.46, 47.83, 43.89, 37.05, 34.48, 31.22, 29.48, 27.51, 26.26, 25.20, 17.25.
(S,E)-4-hydroxy-4-((8S,9S,13S,14S)-3-hydroxy-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-1-(4-(trifluoromethyl)phenyl)pent-1-en-3-one (16b). Following general procedure, F, compound 15b (0.305 g, 0.5 mmol) was deprotected using TBAF (1.55 mL, 1.55 mmol). White powder; 25% yield; 1H NMR (400 MHz, CDCl3) δ 7.74 (d, J = 15.8 Hz, 1H), 7.65–7.52 (m, 4H), 7.04–6.99 (two d, J = 15.8, 8.4 Hz, 2H), 6.54 (dd, J = 8.4, 2.6 Hz, 1H), 6.48 (d, J = 2.6 Hz, 1H), 5.97–5.88 (m, 1H), 4.97 (s, 1H), 4.17 (s, 1H), 2.80–2.71 (m, 2H), 2.24–2.10 (m, 3H), 1.97–1.90 (m, 1H), 1.85–1.77 (m, 2H), 1.68–1.61 (m, 1H), 1.54 (s, 3H), 1.51–1.18 (m, 4H), 0.67 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 200.47, 155.37, 153.43, 142.51, 138.11, 137.67, 132.75, 129.50, 128.71, 126.24, 125.97, 125.94, 121.74, 115.27, 112.68, 79.43, 56.79, 48.12, 43.82, 37.18, 36.17, 31.14, 29.45, 27.54, 26.50, 24.92, 17.53.
(R,E)-4-hydroxy-4-((8S,9S,13S,14S)-3-hydroxy-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-1-(4-(methylthio)phenyl)pent-1-en-3-one (16c). Following general procedure, F, compound 15c (0.294 g, 0.5 mmol) was deprotected using TBAF (1.55 mL, 1.55 mmol). Yellow powder; 25% yield; 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 15.8 Hz, 1H), 7.42 (d, J = 8.4 Hz, 2H), 7.17 (d, J = 8.4 Hz, 2H), 6.98 (d, J = 8.4 Hz, 1H), 6.89 (d, J = 15.8 Hz, 1H), 6.52 (dd, J = 8.4, 2.5 Hz, 1H), 6.47 (d, J = 2.5 Hz, 1H), 5.93 (d, J = 1.7 Hz, 1H), 4.74 (s, 1H), 4.47 (s, 1H), 2.80–2.67 (m, 2H), 2.43 (s, 3H), 2.23–2.16 (m, 1H), 2.10–1.96 (m, 3H), 1.82–1.79 (m, 2H), 1.49 (s, 3H), 1.46–1.42 (m, 1H), 1.34–1.11 (m, 4H), 0.97 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 200.86, 155.41, 153.38, 148.92, 144.56, 143.19, 138.13, 132.83, 129.31, 129.10, 126.22, 125.90, 117.63, 115.23, 112.61, 78.59, 60.50, 57.35, 47.78, 43.86, 37.07, 34.33, 31.18, 29.51, 25.42, 21.09, 17.24, 15.08, 14.21.
(S,E)-4-hydroxy-4-((8S,9S,13S,14S)-3-hydroxy-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-1-(4-(methylthio)phenyl)pent-1-en-3-one (16d). Following general procedure, F, compound 15d (0.294 g, 0.5 mmol) was deprotected using TBAF (1.55 mL, 1.55 mmol). Yellow powder; 71% yield; 1H NMR (600 MHz, CDCl3) δ 7.70 (d, J = 15.7 Hz, 1H), 7.39 (d, J = 8.3 Hz, 2H), 7.14 (d, J = 8.3 Hz, 2H), 7.00 (d, J = 8.5 Hz, 1H), 6.88 (d, J = 15.7 Hz, 1H), 6.54 (dd, J = 8.5, 2.4 Hz, 1H), 6.47 (d, J = 2.4 Hz, 1H), 5.91 (d, J = 1.6 Hz, 1H), 4.43 (s, 1H), 2.78–2.69 (m, 2H), 2.41 (s, 3H), 2.20–2.08 (m, 3H), 1.92–1.86 (m, 2H), 1.80–1.77 (m, 1H), 1.64–1.59 (m, 1H), 1.53 (s, 3H), 1.49–1.15 (m, 4H), 0.65 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 200.69, 155.60, 153.67, 144.21, 143.16, 138.03, 132.59, 130.70, 129.24, 129.05, 126.20, 125.91, 118.30, 115.36, 112.79, 79.28, 56.81, 48.12, 43.82, 37.19, 36.21, 31.14, 29.49, 27.60, 26.53, 25.10, 17.52, 15.08.
(R,E)-4-hydroxy-4-((8S,9S,13S,14S)-3-hydroxy-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-1-(4-methoxyphenyl)pent-1-en-3-one (16e). Following general procedure, F, compound 15e (0.286 g, 0.5 mmol) was deprotected using TBAF (1.55 mL, 1.55 mmol). White powder; 45% yield; 1H NMR (600 MHz, CDCl3) δ 7.75 (d, J = 15.7 Hz, 1H), 7.46 (d, J = 8.8 Hz, 2H), 6.97 (d, J = 8.5 Hz, 1H), 6.84 (d, J = 8.8 Hz, 2H), 6.81 (d, J = 15.7 Hz, 1H), 6.51 (dd, J = 8.8, 2.6 Hz, 1H), 6.46 (d, J = 2.6 Hz, 1H), 5.92 (d, J = 1.8 Hz, 1H), 5.34 (bs, 1H), 4.57 (s, 1H), 3.76 (s, 3H), 2.79–2.66 (m, 2H), 2.21–2.17 (m, 1H), 2.10–1.95 (m, 3H), 1.83–1.76 (m, 2H), 1.49 (s, 3H), 1.47–1.38 (m, 2H), 1.31–1.12 (m, 3H), 0.96 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 200.90, 162.08, 155.48, 153.47, 145.02, 138.10, 132.77, 130.64, 129.23, 127.04, 126.20, 116.34, 115.27, 114.51, 112.66, 78.56, 57.34, 55.49, 47.78, 43.87, 37.09, 34.31, 31.18, 29.52, 27.54, 26.27, 25.47, 17.28.
(R,E)-1-(4-fluorophenyl)-4-hydroxy-4-((8S,9S,13S,14S)-3-hydroxy-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)pent-1-en-3-one (16f). Following general procedure, F, compound 15f (0.280 g, 0.5 mmol) was deprotected using TBAF (1.55 mL, 1.55 mmol). White powder; 58% yield; 1H NMR (600 MHz, CDCl3) δ 7.74 (d, J = 15.8 Hz, 1H), 7.49 (dd, J = 8.7, 5.4 Hz, 2H), 7.01 (t, J = 8.6 Hz, 2H), 6.96 (d, J = 8.5 Hz, 1H), 6.87 (d, J = 15.8 Hz, 1H), 6.51 (dd, J = 8.5, 2.4 Hz, 1H), 6.46 (d, J = 2.4 Hz, 1H), 5.93 (d, J = 1.7 Hz, 1H), 5.34 (s, 1H), 4.50 (s, 1H), 2.79–2.65 (m, 2H), 2.22–2.18 (m, 1H), 2.10–1.96 (m, 3H), 1.83–1.76 (m, 2H), 1.49 (s, 3H), 1.46–1.40 (m, 2H), 1.34–1.10 (m, 3H), 0.96 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 200.85, 155.21, 153.47, 143.94, 138.09, 132.69, 130.80, 130.74, 129.60, 126.19, 118.39, 116.33, 116.19, 115.30, 112.69, 78.74, 57.42, 47.81, 43.89, 37.07, 34.41, 31.21, 29.50, 27.54, 26.27, 25.31, 17.29.
(R,E)-4-hydroxy-4-((8S,9S,13S,14S)-3-hydroxy-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-1-(5-methylthiophen-2-yl)pent-1-en-3-one (16g). Following general procedure, F, compound 15g (0.281 g, 0.5 mmol) was deprotected using TBAF (1.55 mL, 1.55 mmol). Yellow powder; 81% yield; 1H NMR (600 MHz, CDCl3) δ 7.79 (d, J = 15.4 Hz, 1H), 7.09 (d, J = 3.6 Hz, 1H), 6.99 (d, J = 8.4 Hz, 1H), 6.67 (dd, J = 3.6, 1.0 Hz, 1H), 6.57 (d, J = 15.4 Hz, 1H), 6.52 (dd, J = 8.4, 2.6 Hz, 1H), 6.47 (d, J = 2.6 Hz, 1H), 5.90 (dd, J = 3.2, 1.4 Hz, 1H, C-16-H), 4.85 (bs, 1H), 4.48 (s, 1H), 2.82–2.70 (m, 2H), 2.44 (s, 3H), 2.21–2.16 (m, 1H), 2.10–1.96 (m, 3H), 1.83–1.77 (m, 2H), 1.54–1.49 (m, 1H), 1.46 (s, 3H), 1.44–1.39 (m, 1H), 1.33–1.12 (m, 3H), 0.95 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 200.63, 155.39, 153.33, 145.49, 138.17, 137.87, 137.80, 133.69, 132.92, 129.22, 127.05, 126.24, 116.31, 115.23, 112.61, 78.42, 57.33, 47.75, 43.88, 37.08, 34.29, 31.16, 29.53, 27.53, 26.28, 25.49, 17.24, 15.98.
(S,E)-4-hydroxy-4-((8S,9S,13S,14S)-3-hydroxy-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)-1-(5-methylthiophen-2-yl)pent-1-en-3-one (16h). Following general procedure, F, compound 15h (0.281 g, 0.5 mmol) was deprotected using TBAF (1.55 mL, 1.55 mmol). Yellow powder; 25% yield; 1H NMR (600 MHz, CDCl3) δ 7.77 (d, J = 15.3 Hz, 1H), 7.08 (d, J = 3.6 Hz, 1H), 7.04 (d, J = 8.4 Hz, 1H, Ar-H), 6.67 (dd, J = 3.6, 1.1 Hz, 1H), 6.56 (d, J = 15.4 Hz, 1H), 6.54 (dd, J = 8.4, 2.6 Hz, 1H), 6.48 (d, J = 2.6 Hz, 1H), 5.89 (dd, J = 3.3, 1.4 Hz, 1H, C-16-H), 4.87 (bs, 1H), 4.34 (s, 1H), 2.81–2.70 (m, 2H), 2.44 (s, 3H), 2.21–2.12 (m, 3H), 1.95–1.88 (m, 2H), 1.83–1.77 (m, 1H), 1.66–1.60 (m, 1H), 1.50 (s, 3H), 1.41–1.18 (m, 3H), 0.82–0.76 (m, 1H), 0.66 (s, 3H).
(R,E)-1-(5-bromothiophen-2-yl)-4-hydroxy-4-((8S,9S,13S,14S)-3-hydroxy-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)pent-1-en-3-one (16i). Following general procedure, F, compound 15i (0.313 g, 0.5 mmol) was deprotected using TBAF (1.55 mL, 1.55 mmol). Yellow powder; 63% yield; 1H NMR (600 MHz, CDCl3) δ 7.74 (d, J = 15.4 Hz, 1H), 7.01 (d, J = 3.9 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 6.97 (d, J = 3.9 Hz, 1H), 6.60 (d, J = 15.4 Hz, 1H), 6.52 (dd, J = 8.4, 2.7 Hz, 1H), 6.47 (d, J = 2.7 Hz, 1H), 5.91 (dd, J = 3.2, 1.4 Hz, 1H), 5.07 (bs, 1H), 4.41 (s, 1H), 2.80–2.68 (m, 2H), 2.22–2.17 (m, 1H), 2.10–1.96 (m, 3H), 1.82–1.76 (m, 2H), 1.53–1.48 (m, 1H), 1.46 (s, 3H), 1.45–1.40 (m, 1H), 1.35–1.08 (m, 3H), 0.95 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 200.50, 155.09, 153.40, 141.33, 138.15, 136.43, 133.10, 132.78, 131.48, 129.64, 126.20, 117.85, 117.27, 115.27, 112.65, 78.61, 57.40, 47.77, 43.88, 37.07, 34.39, 31.19, 29.51, 27.51, 26.27, 25.34, 17.24.
(R,E)-1-(5-bromofuran-2-yl)-4-hydroxy-4-((8S,9S,13S,14S)-3-hydroxy-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)pent-1-en-3-one (16j). Following general procedure, F, compound 15j (0.305 g, 0.5 mmol) was deprotected using TBAF (1.55 mL, 1.55 mmol). Yellow powder; 45% yield; 1H NMR (600 MHz, CDCl3) δ 7.39 (d, J = 15.5 Hz, 1H), 6.99 (d, J = 8.5 Hz, 1H), 6.74 (d, J = 15.5 Hz, 1H), 6.59 (d, J = 3.5 Hz, 1H), 6.52 (dd, J = 8.5, 2.7 Hz, 1H), 6.47 (d, J = 2.7 Hz, 1H), 6.38 (d, J = 3.5 Hz, 1H), 5.94 (dd, J = 3.3, 1.4 Hz, 1H), 4.88 (bs, 1H), 4.44 (s, 1H), 2.81–2.69 (m, 2H), 2.25–2.20 (m, 1H), 2.10–1.97 (m, 3H), 1.82–1.77 (m, 2H), 1.54–1.50 (m, 1H), 1.48 (s, 3H), 1.47–1.41 (m, 1H), 1.35–1.09 (m, 3H), 0.96 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 200.71, 154.84, 153.34, 153.02, 138.20, 132.91, 129.70, 129.53, 126.50, 126.21, 119.29, 116.65, 115.25, 114.84, 112.61, 78.65, 57.14, 47.76, 43.85, 37.07, 34.42, 31.16, 29.52, 27.51, 26.29, 25.32, 17.26.
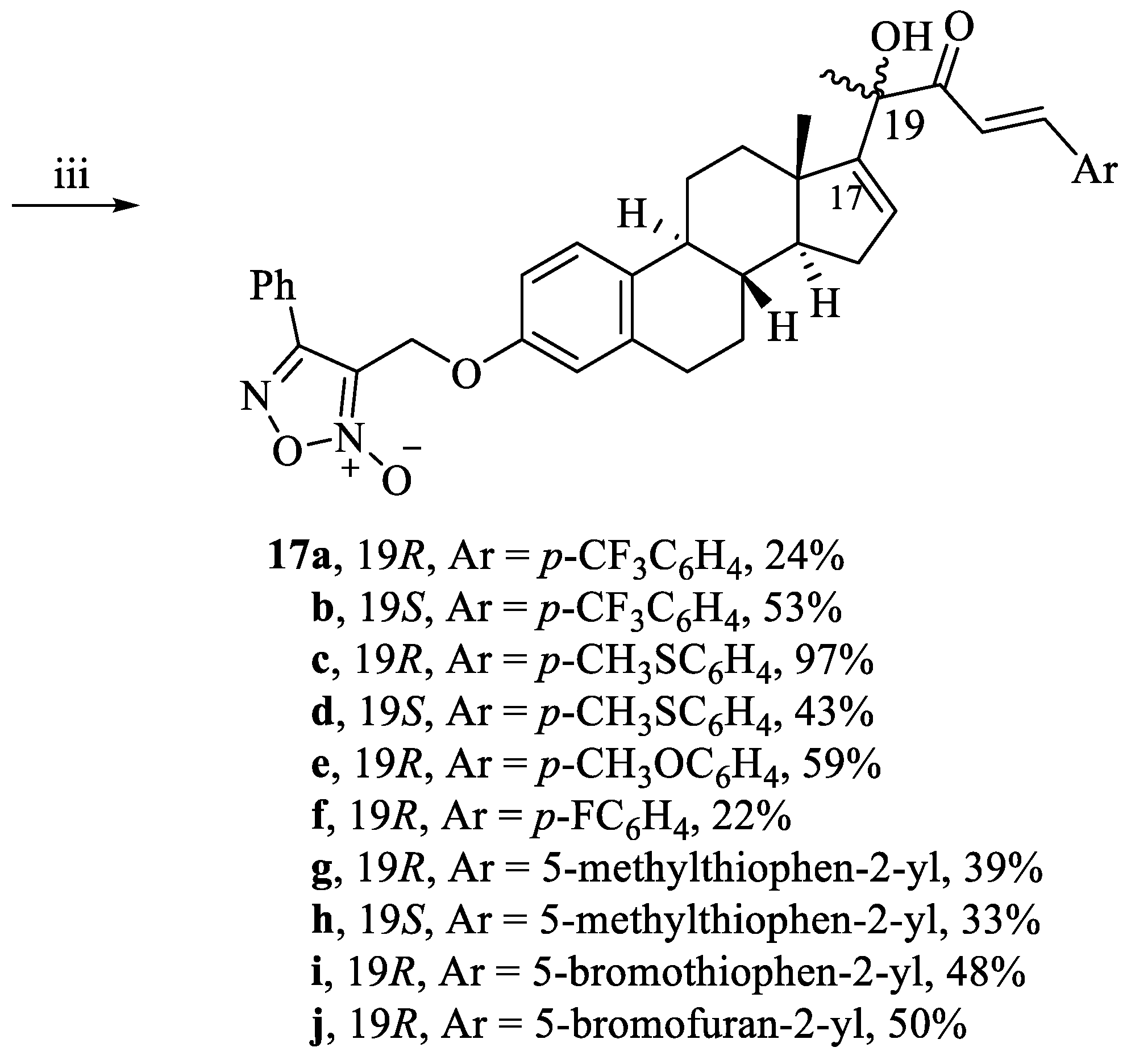
3-((((8S,9S,13S,14S)-17-((R,E)-2-hydroxy-3-oxo-5-(4-(trifluoromethyl)phenyl)pent-4-en-2-yl)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-3-yl)oxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (17a). Following general procedure, G, compound 16a (0.139 g, 0.28 mmol) was alkylated with furoxan mesylate. Yellowish white powder; 24% yield; 1H NMR (600 MHz, CDCl3) δ 7.90–7.88 (m, 3H), 7.73–7.69 (m, 4H), 7.60–7.51 (m, 3H), 7.18 (d, J = 8.7 Hz, 1H), 7.13 (d, J = 15.8 Hz, 1H), 6.79 (dd, J = 8.7, 2.6 Hz, 1H), 6.74 (d, J = 2.6 Hz, 1H), 6.08 (d, J = 1.7 Hz, 1H), 5.08 (s, 2H), 4.44 (s, 1H), 2.95–2.82 (m, 2H), 2.37–2.30 (m, 1H), 2.25–2.18 (m, 1H), 2.17–2.12 (m, 1H), 1.98–1.90 (m, 2H), 1.62 (s, 3H), 1.59–1.53 (m, 2H), 1.45–1.21 (m, 4H), 1.08 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 200.67, 157.16, 155.08, 154.84, 143.00, 138.42, 137.66, 134.62, 131.36, 129.76, 129.35, 128.82, 127.77, 126.40, 126.24, 125.99, 125.97, 121.04, 114.90, 112.24, 78.80, 58.28, 57.42, 47.81, 43.91, 36.94, 34.43, 31.22, 29.61, 27.44, 26.20, 25.25, 17.19. HRESI-MS m/z calcd for [M + Na]+ C39H37F3N2O5: 693.254678, found: 693.257459.
3-((((8S,9S,13S,14S)-17-((S,E)-2-hydroxy-3-oxo-5-(4-(trifluoromethyl)phenyl)pent-4-en-2-yl)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-3-yl)oxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (17b). Following general procedure, G, compound 16b (0.139 g, 0.28 mmol) was alkylated with furoxan mesylate. Yellowish white powder; 53% yield; 1H NMR (600 MHz, CDCl3) δ 7.78 (d, J = 7.0 Hz, 1H), 7.75 (d, J = 15.8 Hz, 1H), 7.61–7.56 (m, 4H), 7.49–7.41 (m, 3H), 7.13 (d, J = 8.6 Hz, 1H), 7.01 (d, J = 15.8 Hz, 1H), 6.71 (dd, J = 8.6, 2.6 Hz, 1H), 6.64 (d, J = 2.6 Hz, 1H), 5.94 (d, J = 1.9 Hz, 1H), 4.98 (s, 2H), 4.11 (s, 1H), 2.85–2.72 (m, 2H), 2.25–2.15 (m, 3H), 1.97–1.91 (m, 2H), 1.85–1.81 (m, 1H), 1.69–1.61 (m, 1H), 1.54 (s, 3H, C20-H3), 1.43–1.15 (m, 3H), 0.81–0.77 (m, 1H), 0.68 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 200.45, 157.15, 155.45, 154.85, 142.44, 138.43, 137.69, 134.66, 131.36, 129.37, 129.35, 128.71, 127.77, 126.47, 126.25, 125.98, 125.95, 121.77, 114.90, 112.28, 112.23, 79.36, 58.29, 56.79, 48.10, 43.88, 37.08, 36.17, 31.14, 29.59, 27.48, 26.46, 24.98, 17.50. HRESI-MS m/z calcd for [M + Na]+: C39H37F3N2O5: 693.254678, found: 693.257450.
3-((((8S,9S,13S,14S)-17-((R,E)-2-hydroxy-5-(4-(methylthio)phenyl)-3-oxopent-4-en-2-yl)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-3-yl)oxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (17c). Following general procedure, G, compound 16c (0.132 g, 0.28 mmol) was alkylated with furoxan mesylate. Yellow powder; 97% yield; 1H NMR (600 MHz, CDCl3) δ 7.80–7.77 (m, 2H), 7.73 (d, J = 15.7 Hz, 1H), 7.48–7.41 (m, 5H), 7.17 (d, J = 8.5 Hz, 2H), 7.07 (d, J = 8.6 Hz, 1H), 6.89 (d, J = 15.7 Hz, 1H), 6.68 (dd, J = 8.6, 2.7 Hz, 1H), 6.63 (d, J = 2.7 Hz, 1H), 5.93 (dd, J = 3.2, 1.4 Hz, 1H), 4.97 (s, 2H), 4.44 (s, 1H), 2.81–2.75 (m, 2H), 2.43 (s, 3H), 2.23–2.19 (m, 1H), 2.13–1.98 (m, 3H), 1.84–1.80 (m, 2H), 1.49 (s, 3H), 1.47–1.44 (m, 1H), 1.35–1.11 (m, 3H), 0.97 (s, 3H), 0.86–0.76 (m, 1H). 13C NMR (151 MHz, CDCl3) δ 200.81, 157.15, 155.45, 154.81, 144.49, 143.19, 138.44, 134.75, 131.34, 130.73, 129.34, 129.22, 129.08, 127.77, 126.43, 126.25, 125.91, 117.65, 114.95, 114.87, 112.23, 78.55, 58.29, 57.32, 47.76, 43.88, 36.96, 34.30, 31.17, 29.64, 27.44, 26.21, 25.45, 17.19, 15.09. HRESI-MS m/z calcd for [M + Na]+: C39H40N2O5S: 671.255014, found: 671.257601.
3-((((8S,9S,13S,14S)-17-((S,E)-2-hydroxy-5-(4-(methylthio)phenyl)-3-oxopent-4-en-2-yl)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-3-yl)oxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (17d). Following general procedure, G, compound 16d (0.132 g, 0.28 mmol) was alkylated with furoxan mesylate. Yellow powder; 43% yield; 1H NMR (600 MHz, CDCl3) δ 7.79–7.76 (m, 2H), 7.69 (d, J = 15.7 Hz, 1H), 7.48–7.42 (m, 3H), 7.40 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 8.4 Hz, 2H), 7.11 (d, J = 8.7 Hz, 1H), 6.89 (d, J = 15.7 Hz, 1H), 6.69 (dd, J = 8.7, 2.7 Hz, 1H), 6.63 (d, J = 2.7 Hz, 1H), 5.91 (d, J = 1.9 Hz, 1H), 4.97 (s, 2H), 4.27 (s, 1H), 2.82–2.74 (m, 2H), 2.42 (s, 3H), 2.22–2.13 (m, 3H), 1.96–1.90 (m, 2H), 1.84–1.81 (m, 1H), 1.65–1.62 (m, 1H), 1.52 (s, 3H), 1.37–1.18 (m, 3H), 0.81–0.77 (m, 1H), 0.66 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 200.64, 157.16, 155.80, 154.85, 143.96, 143.08, 138.45, 134.76, 131.36, 130.76, 129.35, 129.00, 128.93, 127.77, 126.49, 126.26, 125.92, 118.41, 114.90, 112.27, 112.23, 79.12, 58.31, 56.81, 48.09, 43.90, 37.09, 36.21, 31.13, 29.62, 27.51, 26.49, 25.19, 17.46, 15.10. HRESI-MS m/z calcd for [M + Na]+: C39H40N2O5S: 671.255014, found: 671.257434.
3-((((8S,9S,13S,14S)-17-((R,E)-2-hydroxy-5-(4-methoxyphenyl)-3-oxopent-4-en-2-yl)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-3-yl)oxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (17e). Following general procedure, G, compound 16e (0.128 g, 0.28 mmol) was alkylated with furoxan mesylate. Yellow powder; 59% yield; 1H NMR (600 MHz, CDCl3) δ 7.79–7.73 (m, 3H), 7.47 (d, J = 8.4 Hz, 2H), 7.45–7.41 (m, 3H), 7.06 (d, J = 8.6 Hz, 1H), 6.85 (d, J = 8.4 Hz, 2H), 6.82 (d, J = 15.6 Hz, 2H), 6.67 (dd, J = 8.6, 2.6 Hz, 1H), 6.62 (d, J = 2.6 Hz, 1H), 5.92 (d, J = 1.8 Hz, 1H), 4.96 (s, 2H), 4.48 (s, 1H), 3.76 (s, 3H), 2.83–2.70 (m, 2H), 2.22–2.18 (m, 1H), 2.11–1.96 (m, 3H), 1.84–1.80 (m, 2H), 1.54–1.50 (m, 1H), 1.48 (s, 3H), 1.46–1.42 (m, 1H), 1.37–1.13 (m, 2H), 0.96 (s, 3H), 0.82–0.77 (m, 1H). 13C NMR (151 MHz, CDCl3) δ 200.82, 162.07, 157.16, 155.58, 154.81, 144.84, 138.45, 134.79, 131.35, 130.60, 129.35, 129.05, 127.77, 127.05, 126.44, 126.25, 116.39, 114.86, 114.50, 112.22, 78.46, 58.29, 57.29, 55.48, 47.75, 43.88, 36.97, 34.26, 31.17, 29.65, 27.45, 26.22, 25.54, 17.21. HRESI-MS m/z calcd for [M + Na]+: C39H40N2O6: 655.277858, found: 655.279825.
3-((((8S,9S,13S,14S)-17-((R,E)-5-(4-fluorophenyl)-2-hydroxy-3-oxopent-4-en-2-yl)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-3-yl)oxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (17f). Following general procedure, G, compound 16f (0.124 g, 0.28 mmol) was alkylated with furoxan mesylate. Yellow powder; 22% yield; 1H NMR (600 MHz, CDCl3) δ 7.79–7.76 (m, 2H), 7.74 (d, J = 15.8 Hz, 1H), 7.52–7.48 (m, 2H), 7.46–7.41 (m, 3H), 7.06 (d, J = 8.6 Hz, 1H), 7.02 (t, J = 8.6 Hz, 2H), 6.87 (d, J = 15.8 Hz, 1H), 6.67 (dd, J = 8.6, 2.7 Hz, 1H), 6.62 (d, J = 2.7 Hz, 1H), 6.02–5.87 (m, 1H, C-16-H), 4.96 (s, 2H), 4.39 (s, 1H), 2.82–2.71 (m, 2H), 2.23–1.19 (m, 1H), 2.11–1.98 (m, 3H), 1.84–1.80 (m, 2H), 1.55–1.49 (m, 1H), 1.46–1.08 (m, 3H), 0.97 (s, 3H), 0.81–0.78 (m, 1H). 13C NMR (151 MHz, CDCl3) δ 200.74, 165.17, 163.50, 157.14, 155.37, 154.83, 143.72, 138.42, 134.68, 131.34, 130.74, 130.68, 129.34, 127.77, 126.41, 126.27, 118.45, 116.32, 116.17, 114.89, 112.24, 78.61, 58.29, 57.37, 47.79, 43.92, 36.96, 34.35, 31.96, 29.74, 27.46, 22.72, 17.19, 14.15. HRESI-MS m/z calcd for [M + Na]+: C38H37FN2O5: 643.257871, found: 643.259297.
3-((((8S,9S,13S,14S)-17-((R,E)-2-hydroxy-5-(5-methylthiophen-2-yl)-3-oxopent-4-en-2-yl)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-3-yl)oxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (17g). Following general procedure, G, compound 16g (0.120 g, 0.28 mmol) was alkylated with furoxan mesylate. Yellow powder; 39% yield; 1H NMR (600 MHz, CDCl3) δ 7.82–7.76 (m, 3H), 7.49–7.42 (m, 4H), 7.10 (d, J = 3.6 Hz, 1H), 7.07 (d, J = 8.8 Hz, 1H), 6.69–6.65 (m, 2H), 6.63 (d, J = 2.7 Hz, 1H), 6.57 (d, J = 15.4 Hz, 1H), 5.90 (dd, J = 3.6, 1.3 Hz, 1H), 4.97 (s, 2H), 4.43 (s, 1H), 2.84–2.75 (m, 2H), 2.44 (s, 3H), 2.22–2.18 (m, 1H), 2.13–2.03 (m, 2H), 2.02–1.95 (m, 1H), 1.84–1.77 (m, 2H), 1.54–1.49 (m, 1H), 1.46 (s, 3H), 1.37–1.10 (m, 3H), 0.96 (s, 3H), 0.87–0.74 (m, 1H). 13C NMR (151 MHz, CDCl3) δ 200.58, 157.16, 155.43, 154.80, 145.45, 138.47, 137.88, 137.72, 134.80, 133.64, 131.35, 129.35, 129.11, 127.77, 127.05, 126.44, 126.26, 116.34, 114.86, 112.22, 78.37, 58.29, 57.28, 47.72, 43.90, 36.97, 34.24, 31.15, 29.66, 27.45, 26.22, 25.53, 17.19, 15.97. HRESI-MS m/z calcd for [M + Na]+: C37H38N2O5S: 645.239364, found: 645.241095.
3-((((8S,9S,13S,14S)-17-((S,E)-2-hydroxy-5-(5-methylthiophen-2-yl)-3-oxopent-4-en-2-yl)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-3-yl)oxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (17h). Following general procedure, G, compound 16h (0.120 g, 0.28 mmol) was alkylated with furoxan mesylate. Yellow powder; 33% yield; 1H NMR (600 MHz, CDCl3) δ 7.80–7.78 (m, 2H), 7.76 (d, J = 15.3 Hz, 1H), 7.54–7.40 (m, 3H), 7.13 (d, J = 8.6 Hz, 1H), 7.08 (d, J = 3.6 Hz, 1H), 6.70 (dd, J = 8.6, 2.8 Hz, 1H), 6.67 (dd, J = 3.6, 1.1 Hz, 1H), 6.64 (d, J = 2.8 Hz, 1H), 6.57 (d, J = 15.3 Hz, 1H), 5.89 (dd, J = 3.3, 1.4 Hz, 1H), 4.98 (s, 2H), 4.29 (s, 1H), 2.83–2.75 (m, 2H), 2.44 (s, 3H), 2.26–2.14 (m, 3H), 2.00–1.90 (m, 2H), 1.85–1.81 (m, 1H), 1.67–1.62 (m, 1H), 1.50 (s, 3H), 1.42–1.18 (m, 3H), 0.83–0.79 (m, 1H), 0.67 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 207.11, 154.75, 145.27, 138.46, 137.88, 134.80, 133.54, 131.34, 129.34, 128.85, 127.77, 127.01, 126.49, 126.26, 118.66, 117.08, 114.88, 114.88, 112.26, 112.22, 78.81, 66.58, 58.30, 56.78, 48.08, 43.91, 37.08, 36.23, 31.10, 29.62, 27.50, 26.48, 17.40, 15.95. HRESI-MS m/z calcd for [M + Na]+ C37H38N2O5S: 645.239364, found: 645.241750.
3-((((8S,9S,13S,14S)-17-((R,E)-5-(5-bromothiophen-2-yl)-2-hydroxy-3-oxopent-4-en-2-yl)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-3-yl)oxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (17i). Following general procedure, G, compound 16i (0.144 g, 0.28 mmol) was alkylated with furoxan mesylate. Yellowish brown powder; 48% yield; 1H NMR (600 MHz, CDCl3) δ 7.79–7.77 (m, 2H), 7.74 (d, J = 15.4 Hz, 1H), 7.48–7.42 (m, 3H), 7.07 (d, J = 8.6 Hz, 1H), 7.02 (d, J = 3.9 Hz, 1H), 6.97 (d, J = 3.9 Hz, 1H), 6.68 (dd, J = 8.6, 2.7 Hz, 1H), 6.63 (d, J = 2.7 Hz, 1H), 6.60 (d, J = 15.4 Hz, 1H), 5.91 (dd, J = 3.1, 1.3 Hz, 1H), 4.97 (s, 2H), 4.34 (s, 1H), 2.83–2.75 (m, 2H), 2.21–2.19 (m, 1H), 2.12–1.97 (m, 3H), 1.85–1.77 (m, 2H), 1.54–1.51 (m, 1H), 1.46 (s, 3H), 1.38–1.08 (m, 3H), 0.95 (s, 3H), 0.81–0.79 (m, 1H). 13C NMR (151 MHz, CDCl3) δ 200.43, 157.16, 155.16, 154.82, 141.36, 138.47, 136.30, 134.72, 133.02, 131.46, 131.35, 129.49, 129.35, 127.77, 126.42, 126.25, 117.90, 117.20, 114.89, 112.22, 78.52, 58.29, 57.35, 47.75, 43.90, 36.96, 34.34, 31.18, 29.64, 27.43, 26.22, 25.38, 17.17. HRESI-MS m/z calcd for [M + Na]+ C36H35BrN2O5S: 709.134226, found: 709.136204.
3-((((8S,9S,13S,14S)-17-((R,E)-5-(5-bromofuran-2-yl)-2-hydroxy-3-oxopent-4-en-2-yl)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-3-yl)oxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (17j). Following general procedure, G, compound 16j (0.139 g, 0.28 mmol) was alkylated with furoxan mesylate. Yellowish brown powder; 50% yield; 1H NMR (600 MHz, CDCl3) δ 7.80–7.76 (m, 2H), 7.48–7.42 (m, 3H), 7.39 (d, J = 15.4 Hz, 1H), 7.08 (d, J = 8.7 Hz, 1H), 6.74 (d, J = 15.4 Hz, 1H), 6.68 (dd, J = 8.7, 2.7 Hz, 1H), 6.63 (d, J = 2.7 Hz, 1H), 6.60 (d, J = 3.5 Hz, 1H), 6.38 (d, J = 3.5 Hz, 1H), 5.94 (dd, J = 3.2, 1.3 Hz, 1H), 4.98 (s, 2H), 4.38 (s, 1H), 2.84–2.73 (m, 2H), 2.25–2.20 (m, 1H), 2.13–2.06 (m, 2H), 2.02–1.97 (m, 1H), 1.86–1.77 (m, 2H), 1.52–1.49 (m, 1H), 1.48 (s, 3H), 1.37–1.09 (m, 3H), 0.96 (s, 3H), 0.82–0.78 (m, 1H). 13C NMR (151 MHz, CDCl3) δ 200.66, 157.16, 154.88, 154.81, 153.03, 138.50, 134.79, 131.35, 129.60, 129.46, 129.35, 127.77, 126.46, 126.42, 126.26, 119.23, 116.68, 114.89, 114.83, 112.23, 112.21, 78.59, 58.29, 57.10, 47.73, 43.87, 36.97, 34.38, 31.16, 29.66, 27.43, 26.24, 25.36, 17.20. HRESI-MS m/z calcd for [M + Na]+ C36H35BrN2O6: 693.157070, found: 693.159715.
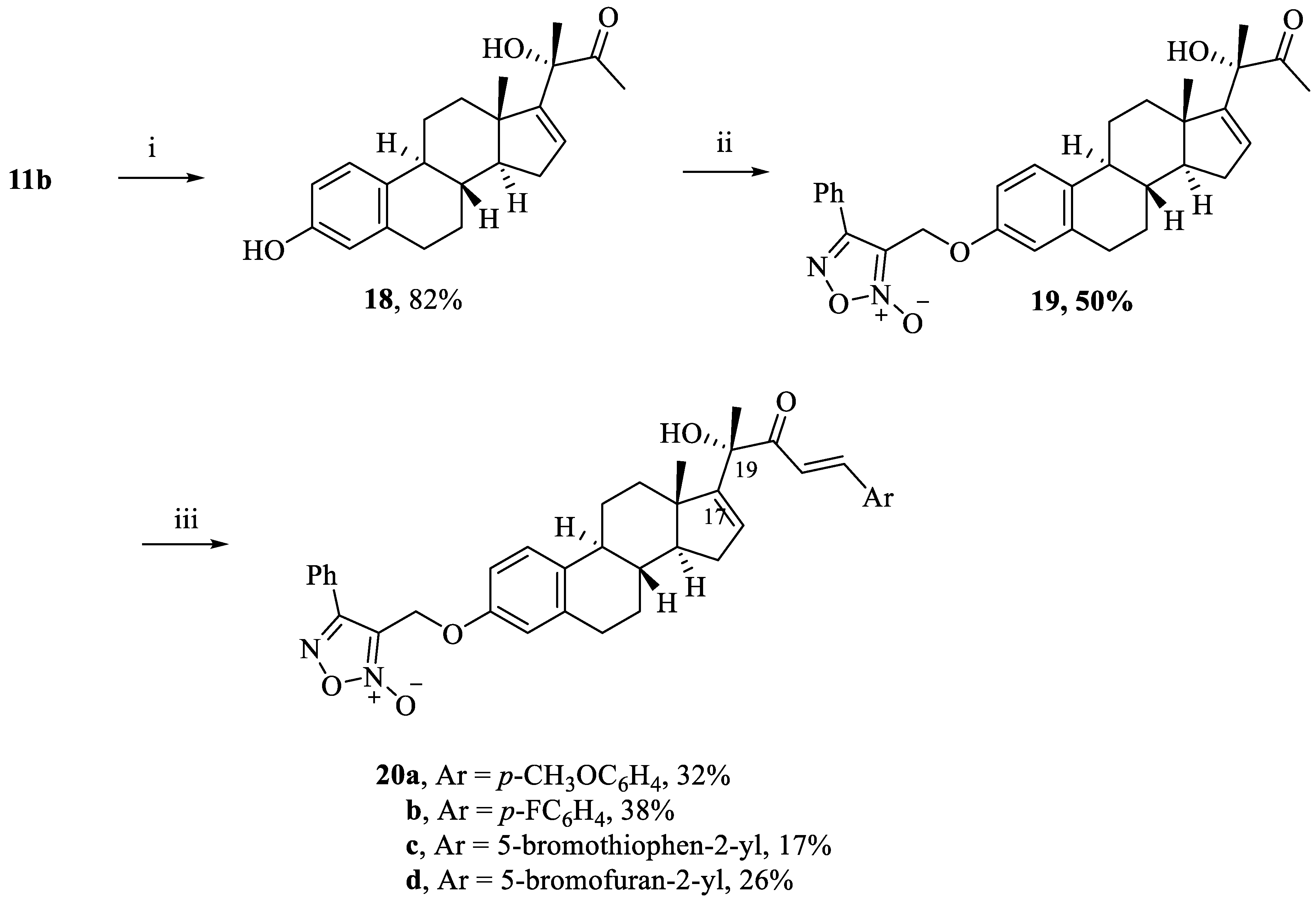
(S)-3-hydroxy-3-((8S,9S,13S,14S)-3-hydroxy-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-17-yl)butan-2-one (18). Following general procedure, F, compound 11b (0.227 g, 0.5 mmol) was deprotected using TBAF (1.55 mL, 1.55 mmol). White powder; 82% yield; 1H NMR (600 MHz, CDCl3) δ 6.99 (d, J = 8.4 Hz, 1H), 6.54 (dd, J = 8.4, 2.7 Hz, 1H), 6.48 (d, J = 2.7 Hz, 1H), 6.10 (s, 1H), 5.83 (dd, J = 3.3, 1.5 Hz, 1H), 4.22 (s, 1H), 2.78–2.67 (m, 2H), 2.18–2.11 (m, 5H), 2.10–2.05 (m, 1H), 1.89–1.86 (m, 1H), 1.80–1.71 (m, 2H), 1.61–1.56 (m, 1H), 1.49 (s, 3H), 1.47–1.17 (m, 3H), 0.82–0.75 (m, 1H), 0.66 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 211.26, 155.17, 153.67, 138.00, 132.51, 129.24, 126.17, 115.40, 112.83, 80.33, 56.63, 48.12, 43.76, 37.16, 36.14, 31.07, 29.49, 27.58, 26.51, 24.78, 23.76, 17.01.
3-((((8S,9S,13S,14S)-17-((S)-2-hydroxy-3-oxobutan-2-yl)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-3-yl)oxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (19). Following general procedure, G, compound 18 (0.095 g, 0.28 mmol) was alkylated with furoxan mesylate. White powder; 50% yield; 1H NMR (600 MHz, CDCl3) δ 7.75–7.72 (m, 2H), 7.42–7.37 (m, 3H), 7.06 (d, J = 8.7 Hz, 1H), 6.65 (dd, J = 8.7, 2.6 Hz, 1H), 6.60 (d, J = 2.6 Hz, 1H), 5.78 (d, J = 1.8 Hz, 1H), 4.91 (s, 2H), 3.98 (s, 1H), 2.79–2.70 (m, 2H), 2.16–2.08 (m, 6H), 1.89–1.85 (m, 2H), 1.81–1.77 (m, 1H), 1.75–1.69 (m, 1H), 1.60–1.55 (m, 1H), 1.43 (s, 3H), 1.36–1.12 (m, 2H), 0.78 - 0.71 (m, 1H), 0.65 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 210.96, 157.13, 155.52, 154.91, 138.40, 134.65, 131.37, 129.36, 128.74, 127.76, 126.45, 126.28, 114.93, 112.27, 112.21, 80.12, 58.32, 56.67, 48.10, 43.90, 37.08, 36.17, 31.06, 29.61, 27.53, 26.50, 24.97, 23.74, 16.96.
3-((((8S,9S,13S,14S)-17-((S,E)-2-hydroxy-5-(4-methoxyphenyl)-3-oxopent-4-en-2-yl)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-3-yl)oxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (20a). Following general procedure, E, compound 11b (0.455 g, 1 mmol) underwent aldol condensation using 4-methoxybenzaldehyde (0.204 g, 1.5 mmol). Yellowish brown powder; 32% yield; 1H NMR (600 MHz, CDCl3) δ 7.80–7.75 (m, 2H), 7.72 (d, J = 15.7 Hz, 1H), 7.48–7.42 (m, 5H), 7.12 (d, J = 8.7 Hz, 1H), 6.84 (d, J = 8.8 Hz, 2H), 6.81 (d, J = 15.7 Hz, 1H), 6.70 (dd, J = 8.7, 2.7 Hz, 1H), 6.63 (d, J = 2.7 Hz, 1H), 5.91 (dd, J = 3.1, 1.2 Hz, 1H), 4.97 (s, 2H), 4.33 (s, 1H), 3.77 (s, 3H), 2.83–2.74 (m, 2H), 2.22–2.16 (m, 3H), 1.94–1.91 (m, 1H), 1.85–1.79 (m, 2H), 1.67–1.62 (m, 1H), 1.52 (s, 3H), 1.48–1.45 (m, 1H), 1.36–1.17 (m, 2H), 0.81–0.79 (m, 1H), 0.67 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 200.66, 162.01, 157.16, 155.91, 154.84, 144.31, 138.45, 134.79, 131.35, 130.49, 129.35, 128.78, 127.77, 127.06, 126.49, 126.26, 117.15, 114.88, 114.48, 112.26, 112.23, 79.01, 58.30, 56.81, 55.47, 48.09, 43.90, 37.09, 36.22, 31.11, 29.62, 27.51, 26.49, 25.26, 17.44. HRESI-MS m/z calcd for [M + Na]+: C39H40N2O6: 655.277858, found: 655.279543.
3-((((8S,9S,13S,14S)-17-((S,E)-5-(4-fluorophenyl)-2-hydroxy-3-oxopent-4-en-2-yl)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-3-yl)oxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (20b). Following general procedure, E, compound 11b (0.455 g, 1 mmol) underwent aldol condensation using 4-fluorobenzaldehyde (0.186 g, 1.5 mmol). White powder; 38% yield; 1H NMR (600 MHz, CDCl3) δ 7.78–7.77 (m, 2H), 7.71 (d, J = 15.8 Hz, 1H), 7.51–7.40 (m, 5H), 7.12 (d, J = 8.7 Hz, 1H), 7.02 (t, J = 8.6 Hz, 2H), 6.87 (d, J = 15.8 Hz, 1H), 6.70 (dd, J = 8.7, 2.7 Hz, 1H), 6.64 (d, J = 2.7 Hz, 1H), 5.97–5.84 (m, 1H, C-16-H), 4.98 (s, 2H), 4.22 (s, 1H), 2.83–2.74 (m, 2H), 2.23–2.14 (m, 3H), 1.94–1.91 (m, 1H), 1.85–1.79 (m, 2H), 1.68–1.64 (m, 1H), 1.52 (s, 3H), 1.49–0.77 (m, 4H), 0.67 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 200.58, 165.13, 163.45, 157.15, 155.69, 154.85, 143.17, 138.44, 134.72, 131.35, 130.63, 130.57, 129.35, 129.06, 127.77, 126.48, 126.26, 119.23, 116.31, 116.16, 114.89, 112.27, 79.18, 58.30, 56.80, 48.10, 43.90, 37.09, 36.20, 31.12, 29.61, 27.50, 26.48, 25.12, 17.46. HRESI-MS m/z calcd for [M + Na]+: C38H37FN2O5: 643.257871, found: 643.259571.
3-((((8S,9S,13S,14S)-17-((S,E)-5-(5-bromothiophen-2-yl)-2-hydroxy-3-oxopent-4-en-2-yl)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-3-yl)oxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (20c). Following general procedure, E, compound 11b (0.455 g, 1 mmol) underwent aldol condensation using 5-bromothiophene-2-carbaldehyde (0.286 g, 1.5 mmol). Yellowish brown powder; 26% yield; 1H NMR (600 MHz, CDCl3) δ 7.80–7.77 (m, 2H), 7.71 (d, J = 15.4 Hz, 1H), 7.49–7.42 (m, 3H), 7.12 (d, J = 8.7 Hz, 1H), 7.01 (d, J = 3.9 Hz, 1H), 6.97 (d, J = 3.9 Hz, 1H), 6.70 (dd, J = 8.7, 2.5 Hz, 1H), 6.64 (d, J = 2.5 Hz, 1H), 6.60 (d, J = 15.4 Hz, 1H), 5.89 (d, J = 1.9 Hz, 1H), 4.98 (s, 2H), 4.17 (s, 1H), 2.82–2.76 (m, 2H), 2.23–2.15 (m, 3H), 1.94–1.90 (m, 1H), 1.83–1.80 (m, 2H), 1.66–1.61 (m, 1H), 1.49 (s, 3H), 1.40–1.16 (m, 3H), 0.84–0.77 (m, 1H), 0.66 (s, 3H). 13C NMR (151 MHz, CDCl3) δ 200.21, 157.15, 155.53, 154.84, 141.37, 138.45, 135.81, 134.73, 132.90, 131.44, 131.35, 129.35, 129.19, 127.77, 126.47, 126.26, 118.65, 117.05, 114.90, 112.26, 112.22, 79.11, 58.30, 56.77, 48.08, 43.89, 37.08, 36.20, 31.12, 29.61, 27.49, 26.47, 25.12, 17.44. HRESI-MS m/z calcd for [M + Na]+ C36H35BrN2O5S: 709.134226, found: 709.136110.
3-((((8S,9S,13S,14S)-17-((S,E)-5-(5-bromofuran-2-yl)-2-hydroxy-3-oxopent-4-en-2-yl)-13-methyl-7,8,9,11,12,13,14,15-octahydro-6H-cyclopenta[a]phenanthren-3-yl)oxy)methyl)-4-phenyl-1,2,5-oxadiazole 2-oxide (20d). Following general procedure, E, compound 11b (0.455 g, 1 mmol) underwent aldol condensation using 5-bromofuran-2-carbaldehyde (0.261 g, 1.5 mmol). Yellowish brown powder; 17% yield; 1H NMR (600 MHz, CDCl3) δ 7.80–7.77 (m, 2H), 7.49–7.42 (m, 3H), 7.36 (d, J = 15.4 Hz, 1H), 7.13 (d, J = 8.7 Hz, 1H), 6.76 (d, J = 15.4 Hz, 1H), 6.70 (dd, J = 8.7, 2.7 Hz, 1H), 6.64 (d, J = 2.7 Hz, 1H), 6.58 (d, J = 3.5 Hz, 1H), 6.37 (d, J = 3.5 Hz, 1H), 5.92 (dd, J = 3.2, 1.4 Hz, 1H), 4.99 (s, 2H), 4.22 (s, 1H), 2.82–2.74 (m, 2H), 2.23–2.14 (m, 3H), 1.98–1.90 (m, 2H), 1.84–1.81 (m, 1H), 1.67–1.62 (m, 1H), 1.52 (s, 3H), 1.49–1.14 (m, 3H), 0.82–0.76 (m, 1H), 0.66 (s, 3H, C-18-H3). 13C NMR (151 MHz, CDCl3) δ 200.56, 157.16, 155.37, 154.84, 153.02, 138.48, 134.78, 131.35, 129.35, 129.24, 128.97, 127.77, 126.48, 126.38, 126.26, 119.07, 117.41, 114.89, 114.77, 112.25, 112.22, 79.15, 58.30, 56.74, 48.08, 43.89, 37.09, 36.25, 31.14, 29.72, 27.50, 26.49, 25.06, 17.44. HRESI-MS m/z calcd for [M + Na]+ C36H35BrN2O6: 693.157070, found: 693.159263.


 ,
, 

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}


