
Chemical Bonding and Dynamic Structural Fluxionality of a Boron-Based Na5B7 Sandwich Cluster

 ,
,  and
and 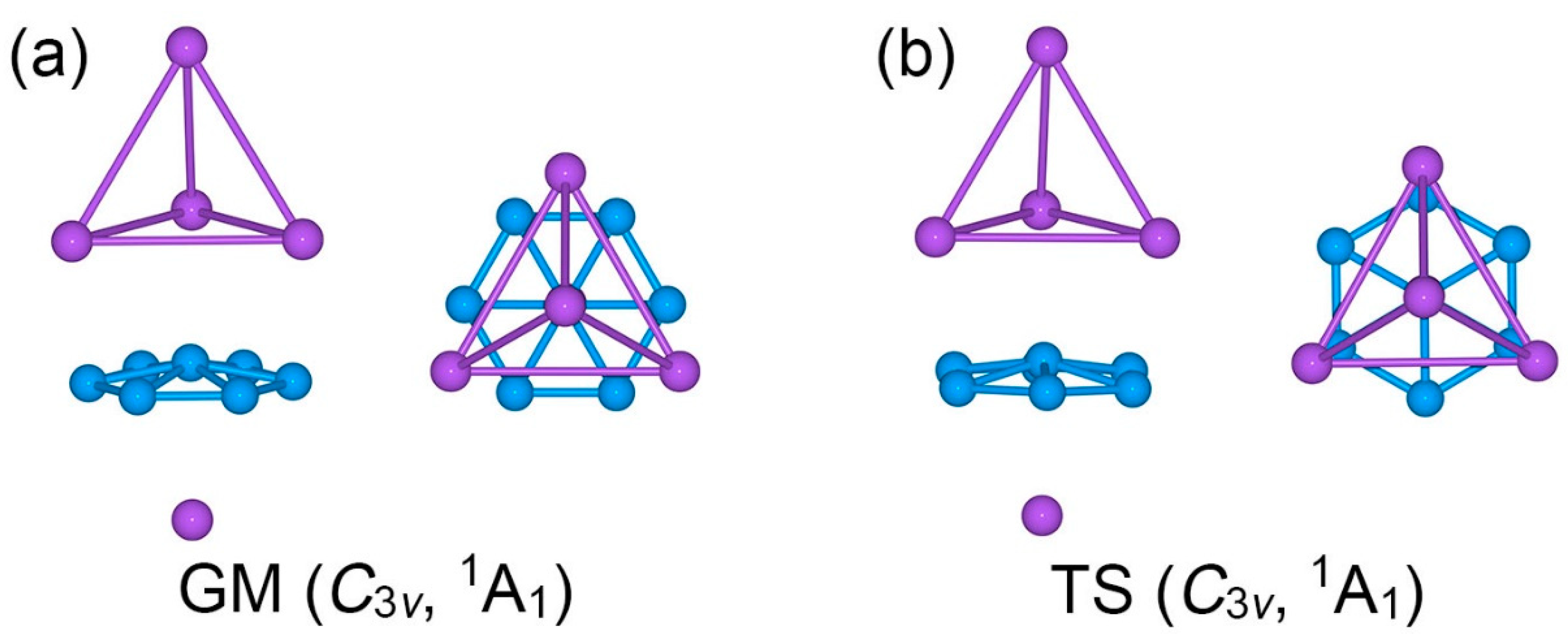{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
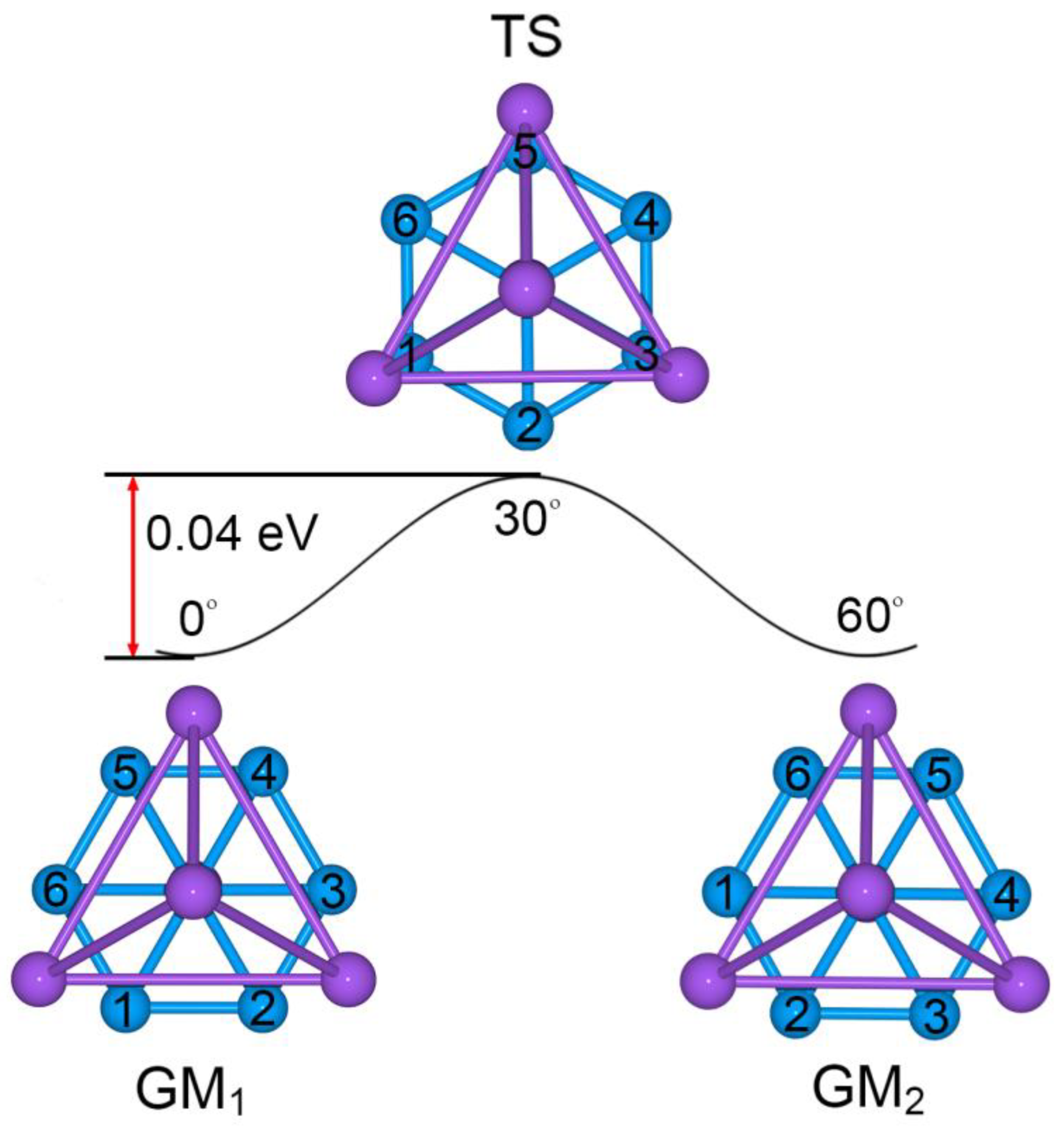
2.1. Global-Minimum and Transition-State Structures
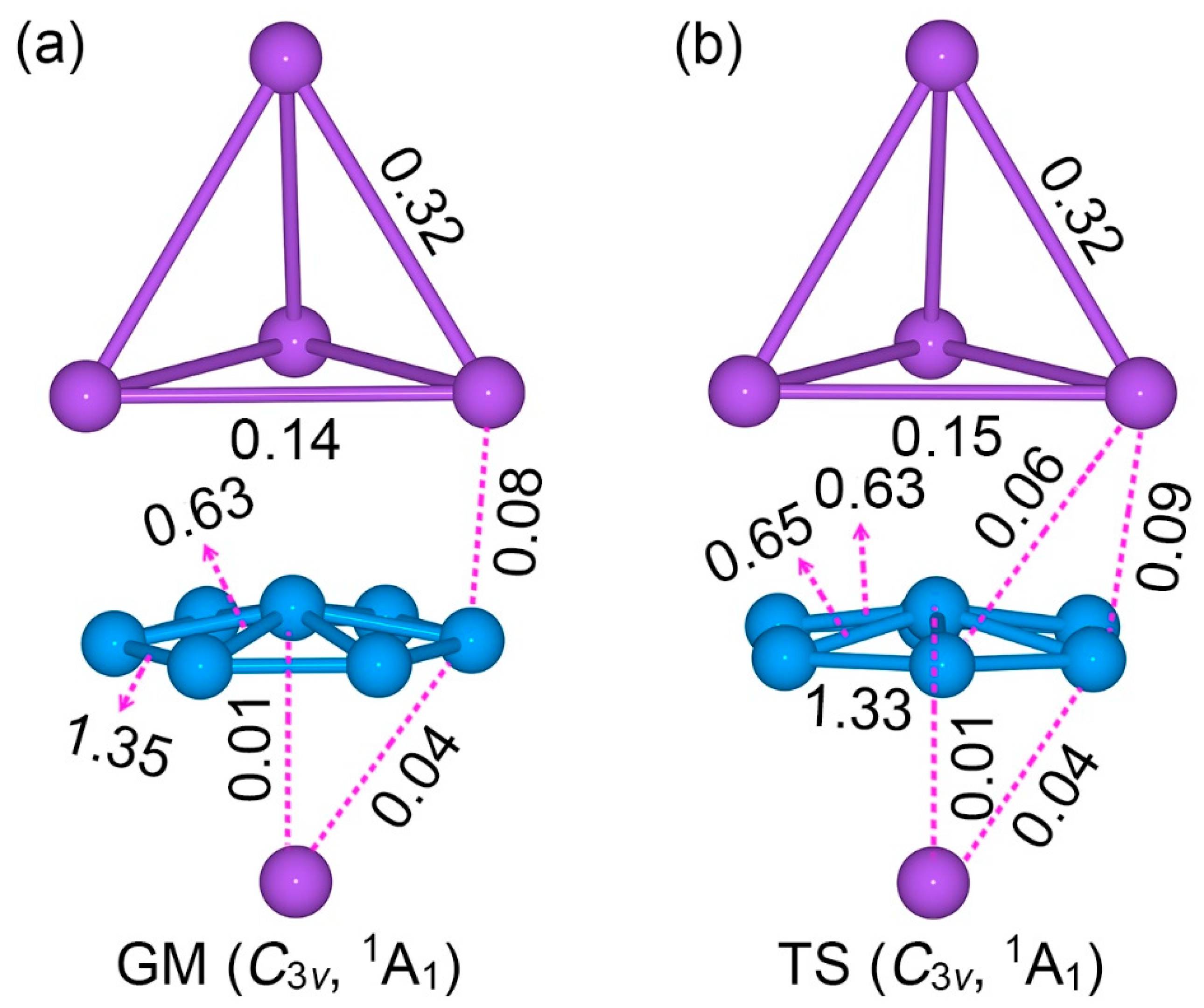
2.2. Bond Distances, Wiberg Bond Indices, and Natural Atomic Charges
3. Methods
4. Discussion
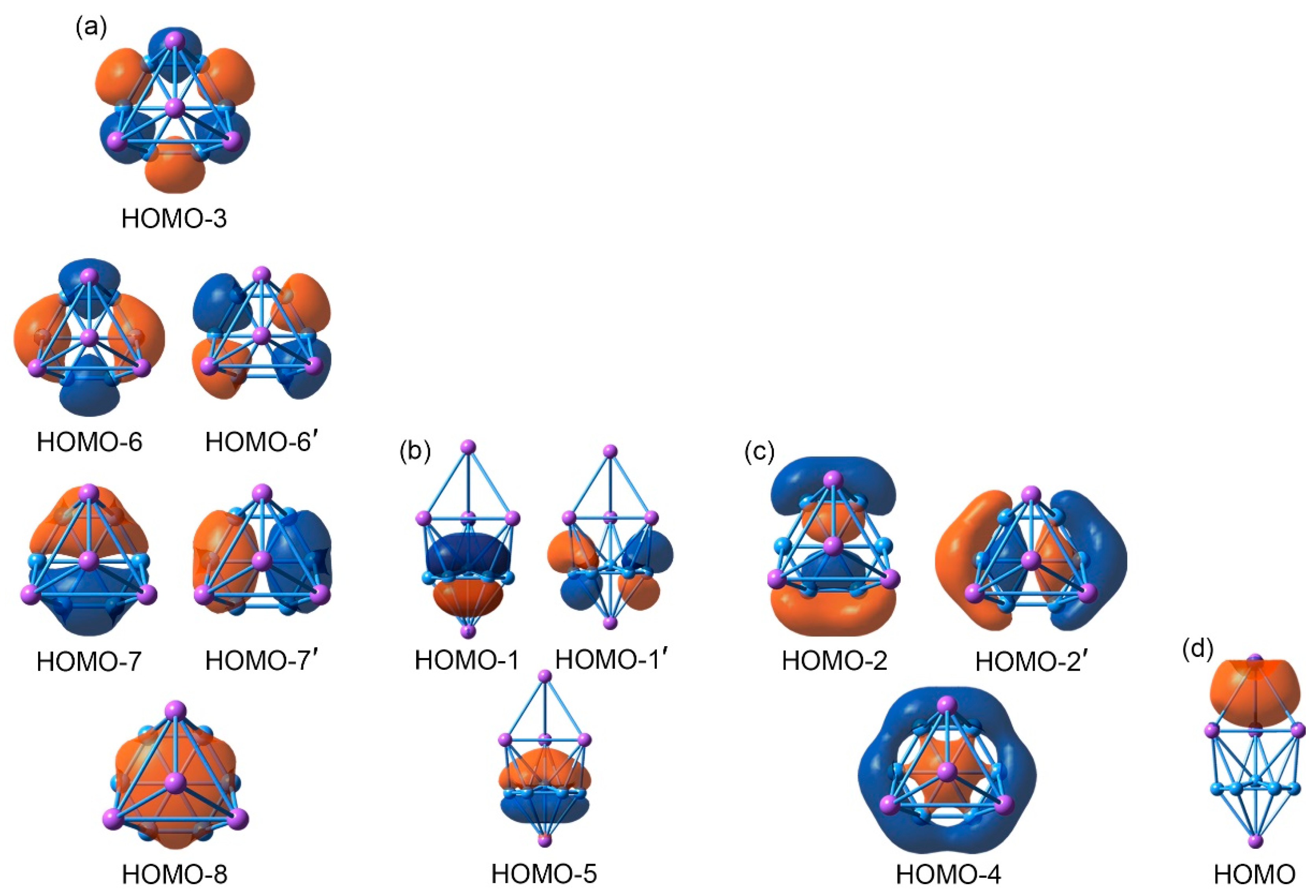
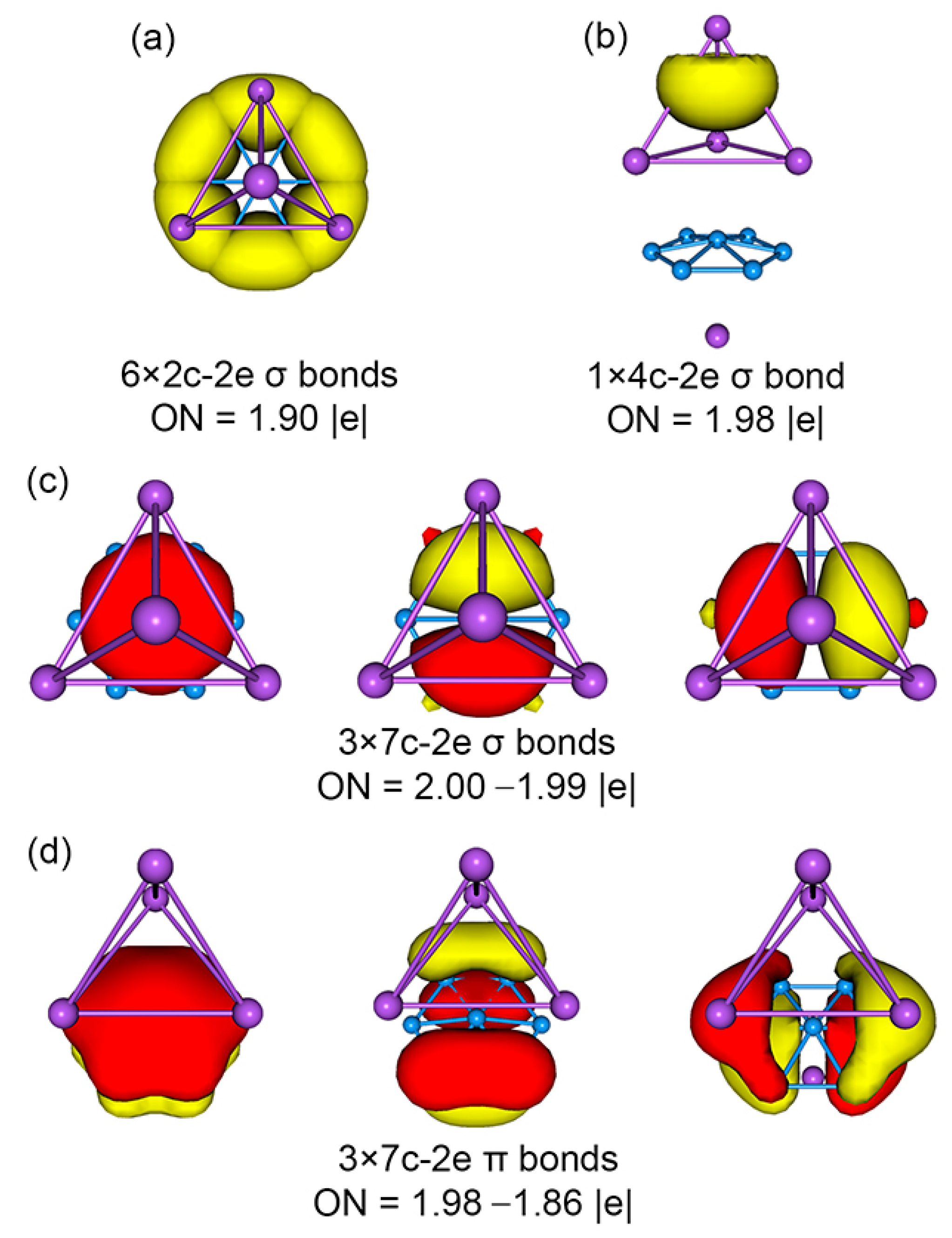
4.1. Chemical Bonding
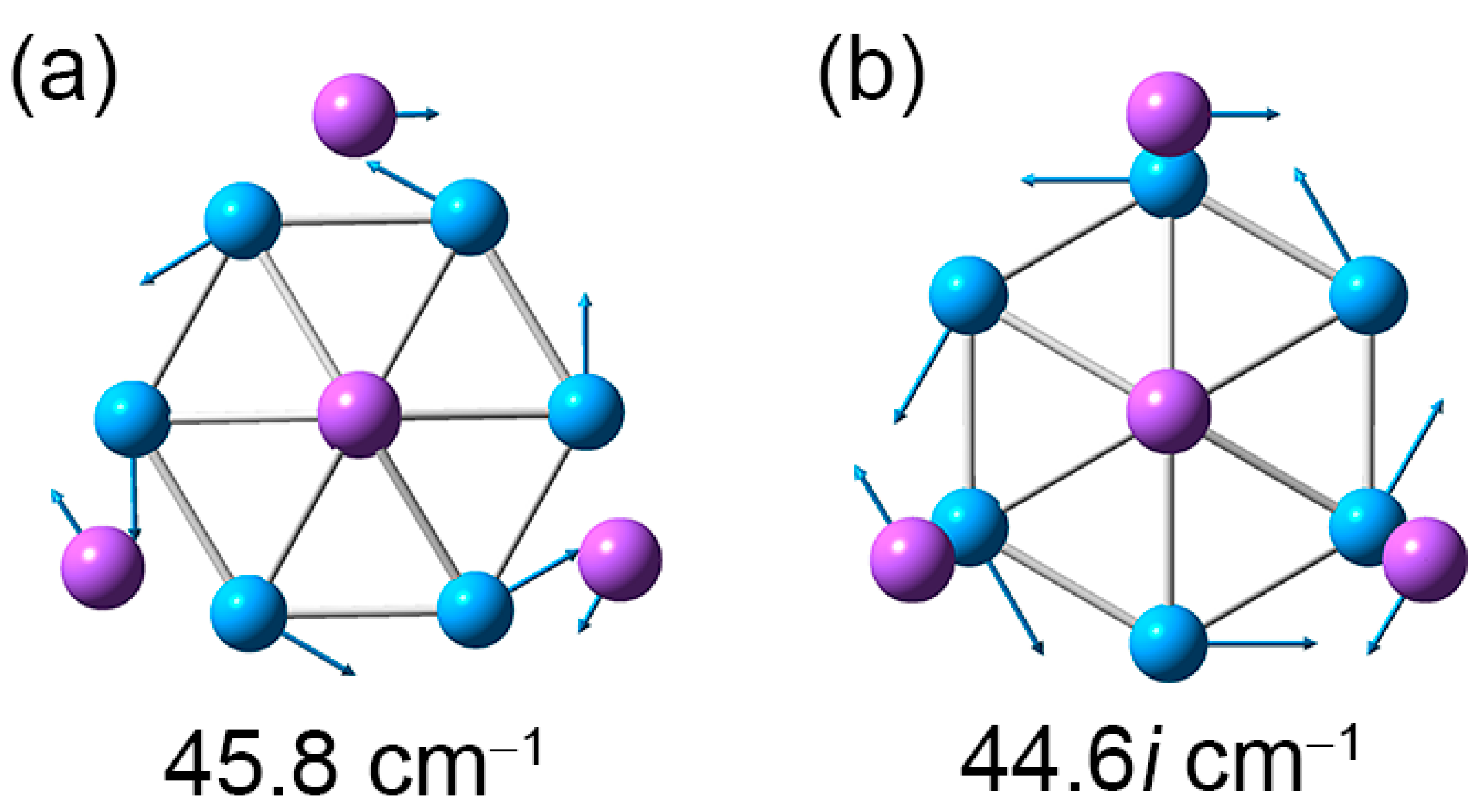
4.2. Dynamic Structural Fluxionality
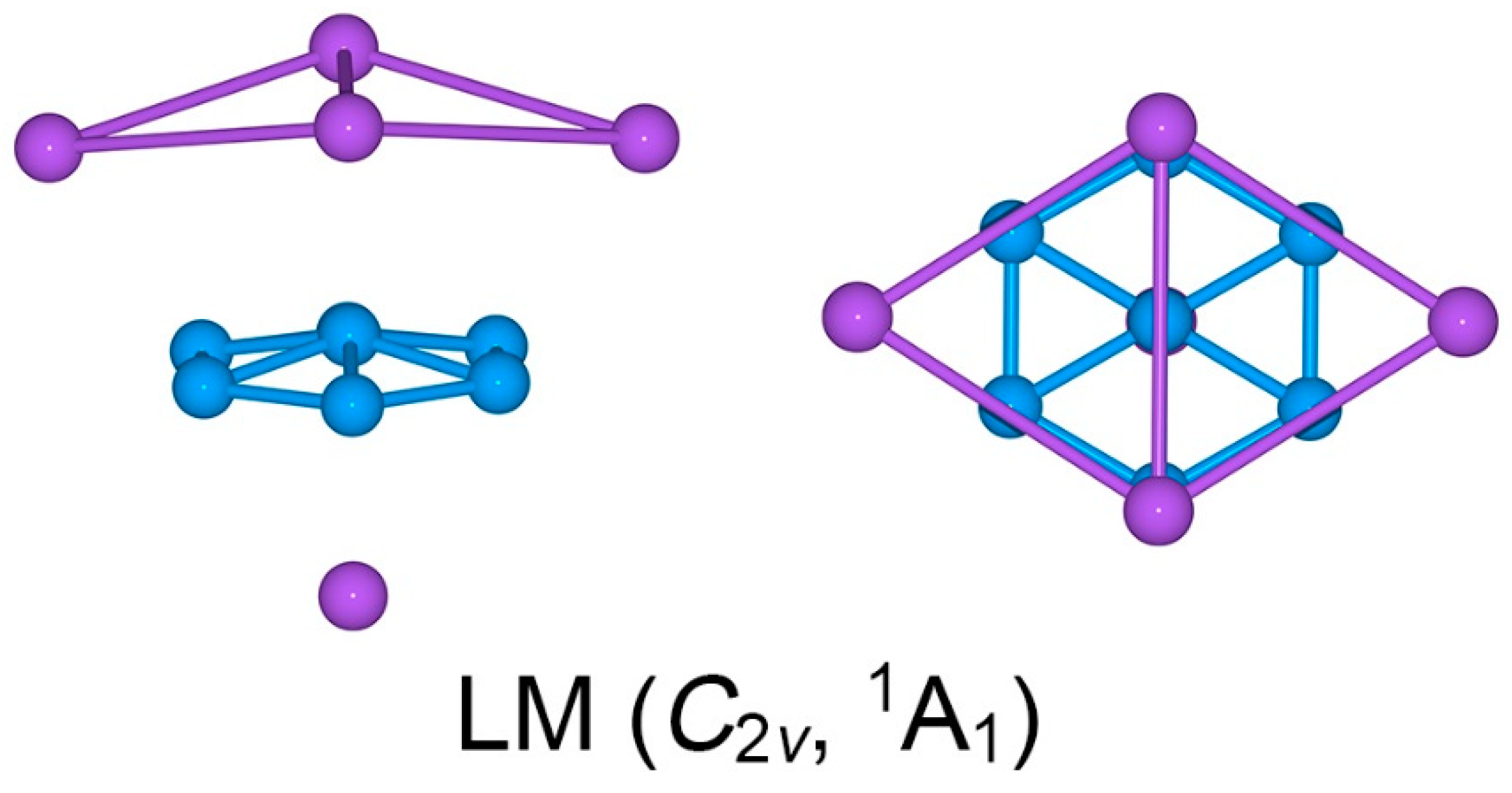
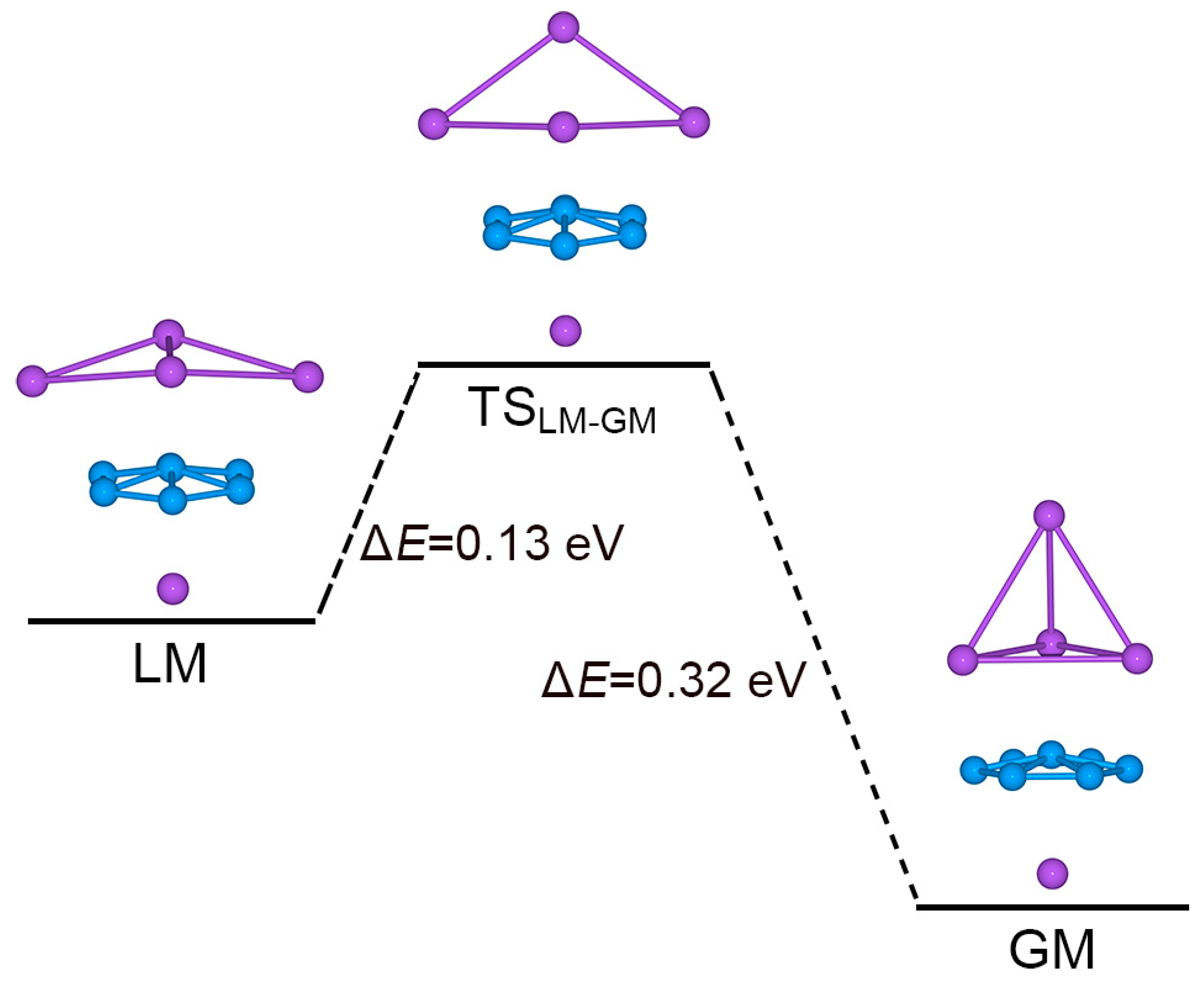
4.3. On the Low-Lying LM Structure: The Importance of Three-Fold π/σ Aromaticity for GM Na5B7 Cluster
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lipscomb, W.N. The Boranes and Their Relatives. Science 1977, 196, 1047–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oger, E.; Crawford, N.R.M.; Kelting, R.; Weis, P.; Kappes, M.M.; Ahlrichs, R. Boron cluster cations: Transition from planar to cylindrical structures. Angew. Chem. Int. Ed. 2007, 46, 8503–8506. [Google Scholar] [CrossRef] [PubMed]
- Zhai, H.-J.; Kiran, B.; Li, J.; Wang, L.-S. Hydrocarbon analogues of boron clusters—Planarity, aromaticity and antiaromaticity. Nat. Mater. 2003, 2, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Zhai, H.-J.; Alexandrova, A.N.; Birch, K.A.; Boldyrev, A.I.; Wang, L.-S. Hepta- and octacoordinate boron in molecular wheels of eight- and nine-atom boron clusters: Observation and confirmation. Angew. Chem. Int. Ed. 2003, 42, 6004–6008. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Zhao, Y.-F.; Li, W.-L.; Jian, T.; Chen, Q.; You, X.-R.; Ou, T.; Zhao, X.-Y.; Zhai, H.-J.; Li, S.-D.; et al. Observation and characterization of the smallest borospherene: B28− and B28. J. Chem. Phys. 2016, 144, 064307. [Google Scholar] [CrossRef] [Green Version]
- Kiran, B.; Bulusu, S.; Zhai, H.-J.; Yoo, S.; Zeng, X.-C.; Wang, L.-S. Photoelectron spectroscopy of aromatic compound clusters of the B12 all-boron benzene: B12Au− and B12(BO)−. Proc. Natl. Acad. Sci. USA 2005, 102, 961–964. [Google Scholar] [CrossRef] [Green Version]
- Li, W.-L.; Chen, Q.; Tian, W.-J.; Bai, H.; Zhao, Y.-F.; Hu, H.-S.; Li, J.; Zhai, H.-J.; Li, S.-D.; Wang, L.-S. The B35 cluster with a double-hexagonal vacancy: A new and more flexible structural motif for borophene. J. Am. Chem. Soc. 2014, 136, 12257–12260. [Google Scholar] [CrossRef]
- Zhai, H.-J.; Zhao, Y.-F.; Li, W.-L.; Chen, Q.; Bai, H.; Hu, H.-S.; Piazza, Z.A.; Tian, W.-J.; Lu, H.-G.; Wu, Y.-B.; et al. Observation of an all-boron fullerene. Nat. Chem. 2014, 6, 727–731. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.-J.; Zhang, J.; Zhong, Q.; Li, W.-B.; Li, S.; Li, H.; Cheng, P.; Meng, S.; Chen, L.; Wu, K.-H. Experimental realization of two-dimensional boron sheets. Nat. Chem. 2016, 8, 563–568. [Google Scholar] [CrossRef] [Green Version]
- Mannix, A.J.; Zhou, X.-F.; Kiraly, B.; Wood, J.D.; Alducin, D.; Myers, B.D.; Liu, X.-L.; Fisher, B.L.; Santiago, U.; Guest, J.R.; et al. Synthesis of borophenes: Anisotropic, two-dimensional boron polymorphs. Science 2015, 350, 1513–1516. [Google Scholar] [CrossRef] [Green Version]
- Sergeeva, A.P.; Zubarev, D.Y.; Zhai, H.-J.; Boldyrev, A.I.; Wang, L.-S. A photoelectron spectroscopic and theoretical study of B16− and B162−: An all-boron naphthalene. J. Am. Chem. Soc. 2008, 130, 7244–7246. [Google Scholar] [CrossRef]
- Alexandrova, A.N.; Boldyrev, A.I.; Zhai, H.-J.; Wang, L.-S. All-boron aromatic clusters as potential new inorganic ligands and building blocks in chemistry. Coord. Chem. Rev. 2006, 250, 2811–2866. [Google Scholar] [CrossRef]
- Gu, F.-L.; Yang, X.-M.; Tang, A.-C.; Jiao, H.-J.; Schleyer, P.V.R. Structure and stability of B13+ clusters. J. Comput. Chem. 1998, 19, 203–214. [Google Scholar] [CrossRef]
- Huang, W.; Sergeeva, A.P.; Zhai, H.-J.; Averkiev, B.B.; Wang, L.-S.; Boldyrev, A.I. A concentric planar doubly π-aromatic B19− cluster. Nat. Chem. 2010, 2, 202–206. [Google Scholar] [CrossRef]
- Jimeénez-Halla, J.O.C.; Islas, R.; Heine, T.; Merino, G. B19−: An aromatic Wankel motor. Angew. Chem. Int. Ed. 2010, 49, 5668–5671. [Google Scholar] [CrossRef]
- Fowler, J.E.; Ugalde, J.M. The curiously stable B13+ cluster and its neutral and anionic counterparts: The advantages of planarity. J. Phys. Chem. A 2000, 104, 397–403. [Google Scholar] [CrossRef]
- Aihara, J.I. B13+ is highly aromatic. J. Phys. Chem. A 2001, 105, 5486–5489. [Google Scholar] [CrossRef]
- Martínez-Guajardo, G.; Sergeeva, A.P.; Boldyrev, A.I.; Heine, T.; Ugalde, J.M.; Merino, G. Unravelling phenomenon of internal rotation in B13+ through chemical bonding analysis. Chem. Commun. 2011, 47, 6242–6244. [Google Scholar] [CrossRef]
- Moreno, D.; Pan, S.; Martínez-Guajardo, G.; Zeonjuk, L.L.; Islas, R.; Osorio, E.; Chattaraj, P.K.; Heine, T.; Merino, G. B182−: A quasi-planar bowl member of the Wankel motor family. Chem. Commun. 2014, 50, 8140–8143. [Google Scholar] [CrossRef]
- Tai, T.-B.; Ceulemans, A.; Nguyen, M.T. Disk aromaticity of the planar and fluxional anionic boron clusters B20−/2−. Chem. Eur. J. 2012, 18, 4510–4512. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Zhao, X.-Y.; Chen, Q.; Zhai, H.-J.; Li, S.-D. B11−: A moving subnanoscale tank tread. Nanoscale 2015, 7, 16054–16060. [Google Scholar] [CrossRef]
- Wang, Y.-J.; You, X.-R.; Chen, Q.; Feng, L.-Y.; Wang, K.; Ou, T.; Zhao, X.-Y.; Zhai, H.-J.; Li, S.-D. Chemical bonding and dynamic fluxionality of a B15+ cluster: A nanoscale double-axle tank tread. Phys. Chem. Chem. Phys. 2016, 18, 15774–15782. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Feng, L.-Y.; Guo, J.-C.; Zhai, H.-J. Dynamic Mg2B8 cluster: A nanoscale compass. Chem. Asian J. 2017, 12, 2899–2903. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.-Y.; Luo, X.-M.; Tian, X.-X.; Lu, H.-G.; Li, S.-D. NiB10, NiB11−, NiB12, and NiB13+: Half-sandwich complexes with the universal coordination bonding pattern of σ plus π double delocalization. J. Clust. Sci. 2019, 30, 115–121. [Google Scholar] [CrossRef]
- Li, W.-L.; Jian, T.; Chen, X.; Li, H.-R.; Chen, T.-T.; Luo, X.-M.; Li, S.-D.; Li, J.; Wang, L.-S. Observation of a metal-centered B2-Ta@B18− tubular molecular rotor and a perfect Ta@B20− boron drum with the record coordination number of twenty. Chem. Commun. 2017, 53, 1587–1590. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Li, H.-R.; Zhao, X.-Y.; Lu, X.-Q.; Mu, Y.-W.; Lu, H.-G.; Li, S.-D. Fluxional bonds in planar B19−, tubular Ta©B20−, and cage-like B39−. J. Comput. Chem. 2019, 40, 966–970. [Google Scholar] [CrossRef] [Green Version]
- Popov, I.A.; Li, W.-L.; Piazza, Z.A.; Boldyrev, A.I.; Wang, L.-S. Complexes between planar boron clusters and transition metals: A photoelectron spectroscopy and ab initio study of CoB12− and RhB12−. J. Phys. Chem. A 2014, 118, 8098–8105. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Moreno, D.; Osorio, E.; Castro, A.C.; Pan, S.; Chattaraj, P.K.; Heine, T.; Merino, G. Structure and bonding of IrB12−: Converting a rigid boron B12 platelet to a Wankel motor. RSC Adv. 2016, 6, 27177–27182. [Google Scholar] [CrossRef]
- Wang, Y.-J.; Feng, L.-Y.; Zhai, H.-J. Divide and stack up: Boron-based sandwich cluster as a subnanoscale propeller. Chem. Asian J. 2019, 14, 2945–2949. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Feng, L.-Y.; Zhai, H.-J. Sandwich-type Na6B7− and Na8B7+ clusters: Charge-transfer complexes, four-fold π/σ aromaticity, and dynamic fluxionality. Phys. Chem. Chem. Phys. 2019, 21, 18338–18345. [Google Scholar] [CrossRef]
- Guo, J.-C.; Feng, L.-Y.; Wang, Y.-J.; Jalife, S.; Vásquez-Espinal, A.; Cabellos, J.L.; Pan, S.; Merino, G.; Zhai, H.-J. Coaxial triple-layered versus helical Be6B11− clusters: Dual structural fluxionality and multifold aromaticity. Angew. Chem. Int. Ed. 2017, 56, 10174–10177. [Google Scholar] [CrossRef]
- Fagiani, M.R.; Song, X.W.; Petkov, P.; Debnath, S.; Gewinner, S.; Schöllkopf, W.; Heine, T.; Fielicke, A.; Asmis, K.R. Structure and fluxionality of B13+ probed by infrared photodissociation spectroscopy. Angew. Chem. Int. Ed. 2017, 56, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Pyykkö, P. Additive covalent radii for single-, double-, and triple-bonded molecules and tetrahedrally bonded crystals: A summary. J. Phys. Chem. A 2015, 119, 2326–2337. [Google Scholar] [CrossRef] [PubMed]
- Alexandrova, A.N.; Boldyrev, A.I.; Zhai, H.-J.; Wang, L.-S. Electronic structure, isomerism, and chemical bonding in B7− and B7. J. Phys. Chem. A 2004, 108, 3509–3517. [Google Scholar] [CrossRef]
- Bonačić-Koutecký, V.; Fantucci, P.; Koutecký, J. Quantum chemistry of small clusters of elements of groups Ia, Ib, and IIa: Fundamental concepts, predictions, and interpretation of experiments. Chem. Rev. 1991, 91, 1035–1108. [Google Scholar] [CrossRef]
- Saunders, M. Stochastic search for isomers on a quantum mechanical surface. J. Comput. Chem. 2004, 25, 621–626. [Google Scholar] [CrossRef]
- Bera, P.P.; Sattelmeyer, K.W.; Saunders, M.; Schaefer III, H.F.; Schleyer, P.v.R. Mindless chemistry. J. Phys. Chem. A 2006, 110, 4287–4290. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. GAUSSIAN 09, Revision D.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Scuseria, G.E.; Janssen, C.L.; Schaefer III, H.F. An efficient reformulation of the closed-shell coupled cluster single and double excitation (CCSD) equations. J. Chem. Phys. 1988, 89, 7382–7387. [Google Scholar] [CrossRef]
- Zubarev, D.Y.; Boldyrev, A.I. Developing paradigms of chemical bonding: Adaptive natural density partitioning. Phys. Chem. Chem. Phys. 2008, 10, 5207–5217. [Google Scholar] [CrossRef] [PubMed]
- Varetto, U. Molekel 5.4.0.8; Swiss National Supercomputing Center: Manno, Switzerland, 2009. [Google Scholar]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. QUICKSTEP: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef] [Green Version]
- Erhardt, S.; Frenking, G.; Chen, Z.-F.; Schleyer, P.v.R. Aromatic boron wheels with more than one carbon atom in the center: C2B8, C3B93+, and C5B11+. Angew. Chem. Int. Ed. 2005, 44, 1078–1082. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Guo, J.-C.; Zhai, H.-J. Why nanoscale tank treads move? Structures, chemical bonding, and molecular dynamics of a doped boron cluster B10C. Nanoscale 2017, 9, 9310–9316. [Google Scholar] [CrossRef] [PubMed]
- Millam, J.M.; Bakken, V.; Chen, W.; Hase, W.L.; Schlegel, H.B. Ab initio classical trajectories on the Born-Oppenheimer surface: Hessian-based integrators using fifth-order polynomial and rational function fits. J. Chem. Phys. 1999, 111, 3800–3805. [Google Scholar] [CrossRef]
- Luo, Z.-X.; Castleman, A.W. Special and general superatoms. Acc. Chem. Res. 2014, 47, 2931–2940. [Google Scholar] [CrossRef] [PubMed]
- Reber, A.C.; Khanna, S.N. Superatoms: Electronic and geometric effects on reactivity. Acc. Chem. Res. 2017, 50, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Jena, P.; Sun, Q. Super atomic clusters: Design rules and potential for building blocks of materials. Chem. Rev. 2018, 118, 5755–5870. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, P.-F.; Wang, Y.-J.; Feng, L.-Y.; Gao, S.-J.; Sun, Q.; Zhai, H.-J. Chemical Bonding and Dynamic Structural Fluxionality of a Boron-Based Na5B7 Sandwich Cluster. Molecules 2023, 28, 3276. https://doi.org/10.3390/molecules28073276
Han P-F, Wang Y-J, Feng L-Y, Gao S-J, Sun Q, Zhai H-J. Chemical Bonding and Dynamic Structural Fluxionality of a Boron-Based Na5B7 Sandwich Cluster. Molecules. 2023; 28(7):3276. https://doi.org/10.3390/molecules28073276
Chicago/Turabian StyleHan, Peng-Fei, Ying-Jin Wang, Lin-Yan Feng, Shu-Juan Gao, Qiang Sun, and Hua-Jin Zhai. 2023. "Chemical Bonding and Dynamic Structural Fluxionality of a Boron-Based Na5B7 Sandwich Cluster" Molecules 28, no. 7: 3276. https://doi.org/10.3390/molecules28073276
APA StyleHan, P. -F., Wang, Y. -J., Feng, L. -Y., Gao, S. -J., Sun, Q., & Zhai, H. -J. (2023). Chemical Bonding and Dynamic Structural Fluxionality of a Boron-Based Na5B7 Sandwich Cluster. Molecules, 28(7), 3276. https://doi.org/10.3390/molecules28073276