
Inhibition of Acetylcholinesterase by Novel Lupinine Derivatives

 , ,
, ,  and
and 
Abstract
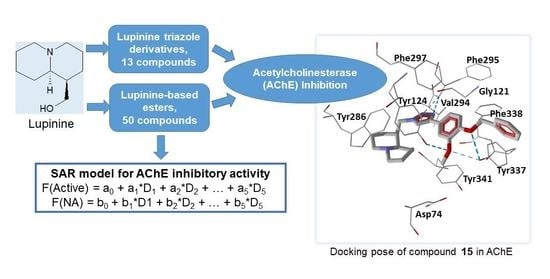
:
1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Results

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Chemical Structure | IC50 (μM) |
|---|---|---|
| Compound 15 |  | 7.2 |
| Compound A |  | 11.8 |
| Compound B |  | 1.2 |
| Compound C |  | 0.6 |
2.3. Molecular Docking
2.4. Classification SAR Model
3. Experimental Section
3.1. Chemistry
3.1.1. General Procedure for Compounds (5–7)
3.1.2. 3-((1-(((1S,9aR)-Octahydro-1H-quinolizine-1-yl)methyl)-1H-1,2,3-triazole-4-yl)methylthio)-1H-1,2,4-triazole-5-amine (5)
3.1.3. (2R,S)-2-(1-(((1S,9aR)-Octahydro-1H-quinolizine-1-yl)methyl)-1H-1,2,3-triazole-4-yl)butane-2,3-diol (6)
3.1.4. 3-Ethoxy-4-((1-(((1S,9aR)-octahydro-1H-quinolizine-1-yl)methyl)-1H-1,2,3-triazole-4-yl)methoxy)benzaldehyde (7)
3.2. Commercial Compounds
3.3. AChE Inhibition Assay
3.4. Cytotoxicity Assay
3.5. Molecular Docking
3.6. Linear Discriminant Analysis (LDA)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Robichaud, A.J. Approaches to palliative therapies for Alzheimer’s disease. Curr. Top. Med. Chem. 2006, 6, 553–568. [Google Scholar] [CrossRef] [PubMed]
- Umar, T.; Meena, R.; Mustehasan; Kumar, P.; Khan, A.A. Recent updates in the development of small molecules as potential clinical candidates for Alzheimer’s disease: A review. Chem. Biol. Drug Des. 2022, 100, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Sepehri, S.; Saeedi, M.; Larijani, B.; Mahdavi, M. Recent developments in the design and synthesis of benzylpyridinium salts: Mimicking donepezil hydrochloride in the treatment of Alzheimer’s disease. Front. Chem. 2022, 10, 936240. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, P.K.; Satheeshkumar, N.; Venkatesh, P.; Venkatesh, M. Lead finding for acetyl cholinesterase inhibitors from natural origin: Structure activity relationship and scope. Mini Rev. Med. Chem. 2011, 11, 247–262. [Google Scholar] [CrossRef]
- Ahn, K. The worldwide trend of using botanical drugs and strategies for developing global drugs. BMB Rep. 2017, 50, 111–116. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.S. Natural products to drugs: Natural product-derived compounds in clinical trials. Nat. Prod. Rep. 2008, 25, 475–516. [Google Scholar] [CrossRef]
- Ayaz, M.; Nawaz, A.; Naz, F.; Ullah, F.; Sadiq, A.; Islam, Z.U. Phytochemicals-based Therapeutics against Alzheimer’s Disease: An Update. Curr. Top. Med. Chem. 2022, 22, 1811–1820. [Google Scholar] [CrossRef]
- Suciati; Poerwantoro, D.; Widyawaruyanti, A.; Ingkaninan, K. Acetylcholinesterase inhibitory activity of extract and fractions from the root of Rauvolfia serpentine (L.) Bth.ex Kurz. J. Basic Clin. Physiol. Pharmacol. 2021, 32, 313–317. [Google Scholar] [CrossRef]
- Tomassini, L.; Ventrone, A.; Frezza, C.; Fabbri, A.M.; Fortuna, S.; Volpe, M.T.; Cometa, M.F. Phytochemical analysis of Viburnum davidii Franch. and cholinesterase inhibitory activity of its dihydrochalcones. Nat. Prod. Res. 2021, 35, 5794–5800. [Google Scholar] [CrossRef]
- Saenkham, A.; Jaratrungtawee, A.; Siriwattanasathien, Y.; Boonsri, P.; Chainok, K.; Suksamrarn, A.; Namsa-Aid, M.; Pattanaprateeb, P.; Suksamrarn, S. Highly potent cholinesterase inhibition of geranylated xanthones from Garcinia fusca and molecular docking studies. Fitoterapia 2020, 146, 104637. [Google Scholar] [CrossRef]
- Hou, Y.; Wang, M.; Sun, C.; Peng, C.; Zhang, Y.; Li, X. Tunicyclin L, a cyclic peptide from Psammosilene tunicoides: Isolation, characterization, conformational studies and biological activity. Fitoterapia 2020, 145, 104628. [Google Scholar] [CrossRef]
- Lou, H.; Yi, P.; Hu, Z.; Li, Y.; Zeng, Y.; Gu, W.; Huang, L.; Yuan, C.; Hao, X. Polycyclic polyprenylated acylphloroglucinols with acetylcholinesterase inhibitory activities from Hypericum perforatum. Fitoterapia 2020, 143, 104550. [Google Scholar] [CrossRef]
- Tuzimski, T.; Petruczynik, A. Determination of Anti-Alzheimer’s Disease Activity of Selected Plant Ingredients. Molecules 2022, 27, 3222. [Google Scholar] [CrossRef]
- Omodeo-Salè, F.; Cortelezzi, L.; Basilico, N.; Casagrande, M.; Sparatore, A.; Taramelli, D. Novel antimalarial aminoquinolines: Heme binding and effects on normal or Plasmodium falciparum-parasitized human erythrocytes. Antimicrob. Agents Chemother. 2009, 53, 4339–4344. [Google Scholar] [CrossRef] [Green Version]
- Van Eijk, J.L.; Radema, M.H. Virgiboidine, A New Alkaloid from Virgilia oroboides and Virgilia divaricata. Planta Med. 1982, 44, 224–226. [Google Scholar] [CrossRef]
- Suzuki, H.; Murakoshi, I.; Saito, K. A novel O-tigloyltransferase for alkaloid biosynthesis in plants. Purification, characterization, and distribution in Lupinus plants. J. Biol. Chem. 1994, 269, 15853–15860. [Google Scholar] [CrossRef]
- Aaron, P.A.; Vu, K.; Gelli, A. An Antivirulence Approach for Preventing Cryptococcus neoformans from Crossing the Blood-Brain Barrier via Novel Natural Product Inhibitors of a Fungal Metalloprotease. mBio 2020, 11, e01249-20. [Google Scholar] [CrossRef]
- Sparatore, A.; Cagnotto, A.; Sparatore, F. Quinolizidinyl derivatives of 2,3-dihydro-2-oxo-1H-benzimidazole-1-carboxylic acid and 1-homolupinanoyl benzimidazolones as ligands for 5-HT3 and 5-HT4 receptors. Farmaco 1999, 54, 248–254. [Google Scholar] [CrossRef]
- Basilico, N.; Parapini, S.; Sparatore, A.; Romeo, S.; Misiano, P.; Vivas, L.; Yardley, V.; Croft, S.L.; Habluetzel, A.; Lucantoni, L.; et al. In vivo and in vitro activities and ADME-tox profile of a quinolizidine-modified 4-aminoquinoline: A potent anti-P. falciparum and anti-P. vivax blood-stage antimalarial. Molecules 2017, 22, 2102. [Google Scholar] [CrossRef] [Green Version]
- Rusconi, C.; Vaiana, N.; Casagrande, M.; Basilico, N.; Parapini, S.; Taramelli, D.; Romeo, S.; Sparatore, A. Synthesis and comparison of antiplasmodial activity of (+), (-) and racemic 7-chloro-4-(N-lupinyl)aminoquinoline. Bioorg. Med. Chem. 2012, 20, 5980–5985. [Google Scholar] [CrossRef]
- Vazzana, I.; Novelli, F.; Sparatore, F.; Sparatore, A.; Fadda, G.; Manca, C. Quinolizidine derivatives with antitubercular activity. Farmaco 1994, 49, 105–110. [Google Scholar] [PubMed]
- Basova, N.E.; Kormilitsyn, B.N.; Perchenok, A.; Rozengart, E.V.; Saakov, V.S.; Suvorov, A.A. Reversible lupininin inhibitors of cholinesterases of mammalian blood and of optical ganglia of individuals of the commander squid Berryteuthis magister from different zones of species areal. Zh. Evol. Biokhim. Fiziol. 2012, 48, 213–218. [Google Scholar] [PubMed]
- Rozengart, E.V. Bisalkaloid derivatives of dicarboxylic acids on the basis of lupinine, anabasine, and cytisine as reversible cholinesterase inhibitors. Dokl. Biochem. Biophys. 2003, 388, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Tasso, B.; Catto, M.; Nicolotti, O.; Novelli, F.; Tonelli, M.; Giangreco, I.; Pisani, L.; Sparatore, A.; Boido, V.; Carotti, A.; et al. Quinolizidinyl derivatives of bi- and tricyclic systems as potent inhibitors of acetyl- and butyrylcholinesterase with potential in Alzheimer’s disease. Eur. J. Med. Chem. 2011, 46, 2170–2184. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, M.; Catto, M.; Tasso, B.; Novelli, F.; Canu, C.; Iusco, G.; Pisani, L.; Stradis, A.D.; Denora, N.; Sparatore, A.; et al. Multitarget Therapeutic Leads for Alzheimer’s Disease: Quinolizidinyl Derivatives of Bi- and Tricyclic Systems as Dual Inhibitors of Cholinesterases and β-Amyloid (Aβ) Aggregation. ChemMedChem 2015, 10, 1040–1053. [Google Scholar] [CrossRef]
- Obaid, R.J.; Naeem, N.; Mughal, E.U.; Al-Rooqi, M.M.; Sadiq, A.; Jassas, R.S.; Moussa, Z.; Ahmed, S.A. Inhibitory potential of nitrogen, oxygen and sulfur containing heterocyclic scaffolds against acetylcholinesterase and butyrylcholinesterase. RSC Adv. 2022, 12, 19764–19855. [Google Scholar] [CrossRef]
- Nurmaganbetov, Z.S.; Nurkenov, O.A.; Fazylov, S.D.; Mukusheva, G.K.; Gazaliev, A.M.; Muldakhmetov, Z.M. Synthesis of 1,2,3-triazolo-quinolizidines based on the quinolizidine alkaloid lupinine. Chem. J. Kazakhstan 2021, 3, 108–118. [Google Scholar] [CrossRef]
- Nurmaganbetov, Z.S.; Savelyev, V.A.; Gatilov, Y.V.; Nurkenov, O.A.; Seidakhmetova, R.B.; Shulgau, Z.T.; Mukusheva, G.K.; Fazylov, S.D.; Shults, E.E. Synthesis and analgesic activity of 1-[(1,2,3-triazol-1-yl)methyl]quinolizines based on the alkaloid lupinine. Chem. Heterocycl. Compd. 2021, 57, 911–919. [Google Scholar] [CrossRef]
- Nurmaganbetov, Z.S.; Fazylov, S.D.; Turdybekov, K.M.; Nurkenov, O.A.; Turdybekov, D.M.; Mukusheva, G.K.; Minayeva, Y.V.; Khabdolda, G. Synthesis and structure of 4-substituted (1S,9aR)-1-[(1,2,3-triazol-1-yl)-methyl]octahydro-1H-quinolysines of lupinine. Bull. Univ. Karaganda Chem. 2022, 2, 12–22. [Google Scholar] [CrossRef]
- Khodja, I.A.; Boulebd, H.; Bensouici, C.; Belfaitah, A. Design, synthesis, biological evaluation, molecular docking, DFT calculations and in silico ADME analysis of (benz)imidazole-hydrazone derivatives as promising antioxidant, antifungal, and anti- acetylcholinesterase agents. J. Mol. Struct. 2020, 1218, 128527. [Google Scholar] [CrossRef]
- Aggarwal, N.; Jain, S.; Chopra, N. Hybrids of Thiazolidin-4-Ones and 1,3,4-Thiadiazole: Synthesis and Biological Screening of A Potential New Class of Acetylcholinesterae Inhibitors. Biointerface Res. Appl. Chem. 2022, 12, 2800–2812. [Google Scholar]
- Mo, J.; Yang, H.; Chen, T.; Li, Q.; Lin, H.; Feng, F.; Liu, W.; Qu, W.; Guo, Q.; Chi, H.; et al. Design, synthesis, biological evaluation, and molecular modeling studies of quinoline-ferulic acid hybrids as cholinesterase inhibitors. Bioorg. Chem. 2019, 93, 103310. [Google Scholar] [CrossRef]
- Lotfi, S.; Rahmani, T.; Hatami, M.; Pouramiri, B.; Kermani, E.T.; Rezvannejad, E.; Mortazavi, M.; Fathi Hafshejani, S.; Askari, N.; Pourjamali, N.; et al. Design, synthesis and biological assessment of acridine derivatives containing 1,3,4-thiadiazole moiety as novel selective acetylcholinesterase inhibitors. Bioorg. Chem. 2020, 105, 104457. [Google Scholar] [CrossRef]
- Catto, M.; Pisani, L.; Leonetti, F.; Nicolotti, O.; Pesce, P.; Stefanachi, A.; Cellamare, S.; Carotti, A. Design, synthesis and biological evaluation of coumarin alkylamines as potent and selective dual binding site inhibitors of acetylcholinesterase. Bioorg. Med. Chem. 2013, 21, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Luz, R.; Almeida, R.B.M.; Albuquerque, M.M.S.; Cerqueira, A.P.M.; Tavares, J.F.; Silva, M.S.D.; Filho, R.B.; Dos Santos Junior, M.C.; Branco, A.; Botura, M.B. Two new dilactonized glycerol glycosides of the dual anticholinesterase active extract from Ocotea daphnifolia using bioguided fractionation and molecular docking studies. Chem. Biol. Drug Des. 2022, 101, 855–864. [Google Scholar] [CrossRef]
- DeLuca, S.; Khar, K.; Meiler, J. Fully Flexible Docking of Medium Sized Ligand Libraries with RosettaLigand. PLoS ONE 2015, 10, e0132508. [Google Scholar] [CrossRef] [PubMed]
- Atanasova, M.; Stavrakov, G.; Philipova, I.; Zheleva, D.; Yordanov, N.; Doytchinova, I. Galantamine derivatives with indole moiety: Docking, design, synthesis and acetylcholinesterase inhibitory activity. Bioorg. Med. Chem. 2015, 23, 5382–5389. [Google Scholar] [CrossRef]
- Mitteroecker, P.; Bookstein, F. Linear Discrimination, Ordination, and the Visualization of Selection Gradients in Modern Morphometrics. Evol. Biol. 2011, 38, 100–114. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Winterfeld, K.; Holschneider, F.W. Über die Konstitution des Lupinins (I. Mitteil). Chem. Ber. Recl. 1931, 64, 137–150. [Google Scholar] [CrossRef]
- Nurkenov, O.A.; Nurmaganbetov, Z.S.; Fazylov, S.D.; Satpaeva, Z.B.; Turdybekov, K.M.; Seilkhanov, T.M.; Talipov, S. Synthesis, Structure, and Properties of New Lupinine O-Acyl Derivatives. Chem. Nat. Compd. 2019, 55, 506–508. [Google Scholar] [CrossRef]
- Lyskov, S.; Chou, F.C.; Conchúir, S.; Der, B.S.; Drew, K.; Kuroda, D.; Xu, J.; Weitzner, B.D.; Renfrew, P.D.; Sripakdeevong, P.; et al. Serverification of molecular modeling applications: The Rosetta Online Server that Includes Everyone (ROSIE). PLoS ONE 2013, 8, e63906. [Google Scholar] [CrossRef] [Green Version]
- Kothiwale, S.; Mendenhall, J.L.; Meiler, J. BCL::Conf: Small molecule conformational sampling using a knowledge based rotamer library. J. Cheminform. 2015, 7, 47. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, G.; Braka, A.; Wu, S. Quantifying functional-group-like structural fragments in molecules and its applications in drug design. J. Chem. Inf. Model. 2023, 63, 2073–2083. [Google Scholar] [CrossRef]





| Compd. | R | IC50 (µM) | Compd. | R | IC50 (µM) |
|---|---|---|---|---|---|
| 5 |  | 101.1 ± 23.9 | 13 |  | 51.8 ± 9.2 |
| 6 |  | N.A. | 14 |  | N.A. |
| 7 |  | 73.4 ± 16.7 | 15 |  | 7.2 ± 0.2 |
| 8 | ---CH2OH | N.A. | 16 |  | 141.2 ± 15.3 |
| 9 | ---C(CH3)2OH | N.A. | |||
| 10 | ---C(CH3)2O(CH2)2CN | N.A. | |||
| 11 |  | N.A. | |||
| 12 |  | N.A. | 17 |  | 68.1 ± 2.2 |

| Compd. | R | IC50 (µM) | Compd. | R | IC50 (µM) |
|---|---|---|---|---|---|
| 22 | ---(CH2)3Cl | 39.5 ± 7.2 | 44 |  | 68.9 ± 5.8 |
| 25 |  | 24.4 ± 3.4 | |||
| 41 |  | 99.4 ± 12.3 | 49 |  | 111.3 ± 12.5 |
| 43 |  | 89.6 ± 7.7 | 64 |  | 74.1 ± 6.8 |
| Descriptor | Coefficient of Classification Functions | ||
|---|---|---|---|
| Active | NA | ||
| Intercept | −61.838 | −51.489 | |
| D1 | MW | 0.064 | 0.046 |
| D2 | Nrot | −0.848 | −0.133 |
| D3 | MR | 1.450 | 1.354 |
| D4 | sLogS | 9.175 | 8.580 |
| D5 | Q | −7.863 | −5.265 |
| Percent | Non-Active (Calculated) | Active (Calculated) | |
|---|---|---|---|
| Non-active (observed) | 81.4 | 35 | 8 |
| Active (observed) | 85.7 | 1 | 6 |
| Total | 82.0 | 36 | 14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schepetkin, I.A.; Nurmaganbetov, Z.S.; Fazylov, S.D.; Nurkenov, O.A.; Khlebnikov, A.I.; Seilkhanov, T.M.; Kishkentaeva, A.S.; Shults, E.E.; Quinn, M.T. Inhibition of Acetylcholinesterase by Novel Lupinine Derivatives. Molecules 2023, 28, 3357. https://doi.org/10.3390/molecules28083357
Schepetkin IA, Nurmaganbetov ZS, Fazylov SD, Nurkenov OA, Khlebnikov AI, Seilkhanov TM, Kishkentaeva AS, Shults EE, Quinn MT. Inhibition of Acetylcholinesterase by Novel Lupinine Derivatives. Molecules. 2023; 28(8):3357. https://doi.org/10.3390/molecules28083357
Chicago/Turabian StyleSchepetkin, Igor A., Zhangeldy S. Nurmaganbetov, Serik D. Fazylov, Oralgazy A. Nurkenov, Andrei I. Khlebnikov, Tulegen M. Seilkhanov, Anarkul S. Kishkentaeva, Elvira E. Shults, and Mark T. Quinn. 2023. "Inhibition of Acetylcholinesterase by Novel Lupinine Derivatives" Molecules 28, no. 8: 3357. https://doi.org/10.3390/molecules28083357
APA StyleSchepetkin, I. A., Nurmaganbetov, Z. S., Fazylov, S. D., Nurkenov, O. A., Khlebnikov, A. I., Seilkhanov, T. M., Kishkentaeva, A. S., Shults, E. E., & Quinn, M. T. (2023). Inhibition of Acetylcholinesterase by Novel Lupinine Derivatives. Molecules, 28(8), 3357. https://doi.org/10.3390/molecules28083357