
The Dichotomy of Mn–H Bond Cleavage and Kinetic Hydricity of Tricarbonyl Manganese Hydride Complexes

 , ,
, ,  ,
,  ,
,  and
and
Abstract
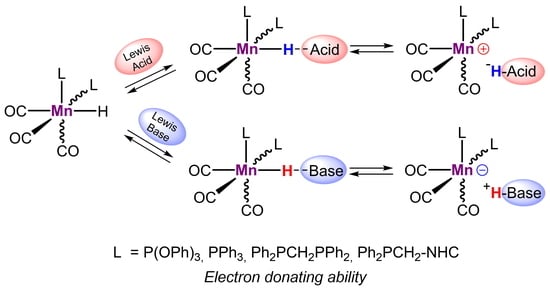
:
1. Introduction
2. Results and Discussion
2.1. Interaction of Tricarbonyl Manganese Hydrides 1–4 with Bases
2.2. Interaction of Tricarbonyl Manganese Hydrides with Lewis Acids
2.3. Kinetic Hydricity of Manganese Tricarbonyl Complexes
3. Materials and Methods
- Synthesis of fac–[(P–NHC)Mn(CO)3(MeCN)]BF4 (4MeCN)
- Synthesis of mer,trans–[(PPh3)2Mn(CO)3(CH3CN)][BF4] (2MeCN)
3.1. General Procedure for the Interaction with Bases
- For Variable Temperature IR Studies
- For Variable Temperature NMR Studies
3.2. General Procedure for the Interaction with Lewis Acid
- For Variable Temperature NMR Studies
- For Variable Temperature IR Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Valyaev, D.A.; Lavigne, G.; Lugan, N. Manganese organometallic compounds in homogeneous catalysis: Past, present, and prospects. Coord. Chem. Rev. 2016, 308, 191–235. [Google Scholar] [CrossRef]
- Mukherjee, A.; Milstein, D. Homogeneous catalysis by cobalt and manganese pincer complexes. ACS Catal. 2018, 8, 11435–11469. [Google Scholar] [CrossRef]
- Gulyaeva, E.S.; Osipova, E.S.; Buhaibeh, R.; Canac, Y.; Sortais, J.-B.; Valyaev, D.A. Towards ligand simplification in manganese-catalyzed hydrogenation and hydrosilylation processes. Coord. Chem. Rev. 2022, 458, 214421. [Google Scholar] [CrossRef]
- Buhaibeh, R.; Filippov, O.A.; Bruneau-Voisine, A.; Willot, J.; Duhayon, C.; Valyaev, D.A.; Lugan, N.; Canac, Y.; Sortais, J.B. Phosphine-NHC manganese hydrogenation catalyst exhibiting a non-classical metal-ligand cooperative H2 activation mode. Angew. Chem. Int. Ed. 2019, 131, 6799–6803. [Google Scholar] [CrossRef] [Green Version]
- Kireev, N.V.; Filippov, O.A.; Gulyaeva, E.S.; Shubina, E.S.; Vendier, L.; Canac, Y.; Sortais, J.-B.; Lugan, N.; Valyaev, D.A. Bis[diphenylphosphino]methane and its bridge-substituted analogues as chemically non-innocent ligands for H2 activation. Chem. Commun. 2020, 56, 2139–2142. [Google Scholar] [CrossRef]
- Pearson, R.G. The transition-metal-hydrogen bond. Chem. Rev. 1985, 85, 41–49. [Google Scholar] [CrossRef]
- Crabtree, R.H. The Organometallic Chemistry of the Transition Metals; John Wiley & Sons: Hoboken, NJ, USA, 2009. [Google Scholar]
- Kristjánsdóttir, S.; Norton, J. Transition Metal Hydrides; Dedieu, A., Ed.; VCH: New York, NY, USA, 1992; Volume 309. [Google Scholar]
- Bullock, R.M. Catalytic ionic hydrogenations. Chem. Eur. J. 2004, 10, 2366–2374. [Google Scholar] [CrossRef]
- Jordan, R.F.; Norton, J.R. Kinetic and thermodynamic acidity of hydrido transition-metal complexes. 1. Periodic trends in Group VI complexes and substituent effects in osmium complexes. J. Am. Chem. Soc. 1982, 104, 1255–1263. [Google Scholar] [CrossRef]
- Cheng, T.-Y.; Brunschwig, B.S.; Bullock, R.M. Hydride transfer reactions of transition metal hydrides: Kinetic hydricity of metal carbonyl hydrides. J. Am. Chem. Soc. 1998, 120, 13121–13137. [Google Scholar] [CrossRef]
- Bullock, R.M. Proton transfer from metal hydrides to metal alkynyl complexes. Remarkable carbon basicity of (C5H5)(PMe3)2Ru-CC-CMe3. J. Am. Chem. Soc. 1987, 109, 8087–8089. [Google Scholar] [CrossRef]
- Filippov, O.; Golub, I.; Osipova, E.; Kirkina, V.; Gutsul, E.; Belkova, N. Activation of M—H bond upon the complexation of transition metal hydrides with acids and bases. Russ. Chem. Bull. 2014, 63, 2428–2433. [Google Scholar] [CrossRef]
- Belkova, N.V.; Filippov, O.A.; Osipova, E.S.; Safronov, S.V.; Epstein, L.M.; Shubina, E.S. Influence of phosphine (pincer) ligands on the transition metal hydrides reactivity. Coord. Chem. Rev. 2021, 438, 213799. [Google Scholar] [CrossRef]
- Wiedner, E.S.; Chambers, M.B.; Pitman, C.L.; Bullock, R.M.; Miller, A.J.; Appel, A.M. Thermodynamic hydricity of transition metal hydrides. Chem. Rev. 2016, 116, 8655–8692. [Google Scholar] [CrossRef]
- Edidin, R.T.; Sullivan, J.M.; Norton, J.R. Kinetic and thermodynamic acidity of hydrido transition-metal complexes. 4. Kinetic acidities toward aniline and their use in identifying proton-transfer mechanisms. J. Am. Chem. Soc. 1987, 109, 3945–3953. [Google Scholar] [CrossRef]
- Sandhya, K.; Suresh, C.H. Quantification of thermodynamic hydridicity of hydride complexes of Mn, Re, Mo, and W using the molecular electrostatic potential. J. Phys. Chem. 2017, 121, 2814–2819. [Google Scholar] [CrossRef]
- Kuchynka, D.; Amatore, C.; Kochi, J. Electrooxidation of metal cabonyl anions. Formation and reactivity of 17-electron manganese (0) radicals. J. Organomet. Chem. 1987, 328, 133–154. [Google Scholar] [CrossRef]
- Belkova, N.V.; Epstein, L.M.; Filippov, O.A.; Shubina, E.S. Hydrogen and dihydrogen bonds in the reactions of metal hydrides. Chem. Rev. 2016, 116, 8545–8587. [Google Scholar] [CrossRef]
- Belkova, N.V.; Epstein, L.M.; Shubina, E.S. Dihydrogen bonding, proton transfer and beyond: What we can learn from kinetics and thermodynamics. Eur. J. Inorg. Chem. 2010, 23, 3555–3565. [Google Scholar] [CrossRef]
- Ault, B.S.; Becker, T.M.; Li, G.Q.; Orchin, M. The infrared spectra and theoretical calculations of frequencies of fac-tricarbonyl octahedral complexes of manganese (I). Spectrochim. Acta A Mol. Biomol. Spectrosc. 2004, 60, 2567–2572. [Google Scholar] [CrossRef]
- Belkova, N.V.; Gutsul, E.I.; Filippov, O.A.; Levina, V.A.; Valyaev, D.A.; Epstein, L.M.; Lledos, A.; Shubina, E.S. Intermolecular hydrogen bonding between neutral transition metal hydrides (η5-C5H5)M(CO)3H (M = Mo, W) and bases. J. Am. Chem. Soc. 2006, 128, 3486–3487. [Google Scholar] [CrossRef] [PubMed]
- Levina, V.A.; Filippov, O.A.; Gutsul, E.I.; Belkova, N.V.; Epstein, L.M.; Lledos, A.; Shubina, E.S. Neutral transition metal hydrides as acids in hydrogen bonding and proton transfer: Media polarity and specific solvation effects. J. Am. Chem. Soc. 2010, 132, 11234–11246. [Google Scholar] [CrossRef]
- Levina, V.A.; Rossin, A.; Belkova, N.V.; Chierotti, M.R.; Epstein, L.M.; Filippov, O.A.; Gobetto, R.; Gonsalvi, L.; Lledós, A.; Shubina, E.S. Acid–base interaction between transition-metal hydrides: Dihydrogen bonding and dihydrogen evolution. Angew. Chem. Int. Ed. 2011, 123, 1403–1406. [Google Scholar] [CrossRef]
- Osipova, E.S.; Belkova, N.V.; Epstein, L.M.; Filippov, O.A.; Kirkina, V.A.; Titova, E.M.; Rossin, A.; Peruzzini, M.; Shubina, E.S. Dihydrogen bonding and proton transfer from MH and OH acids to Group 10 metal hydrides [(tBuPCP)MH][tBuPCP = κ3-2,6-(tBu2PCH2)2C6H3; M = Ni, Pd]. Eur. J. Inorg. Chem. 2016, 2016, 1415–1424. [Google Scholar] [CrossRef]
- Eckert, F.; Leito, I.; Kaljurand, I.; Kütt, A.; Klamt, A.; Diedenhofen, M. Prediction of acidity in acetonitrile solution with COSMO-RS. J. Comput. Chem. 2009, 30, 799–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osipova, E.S.; Gulyaeva, E.S.; Kireev, N.V.; Kovalenko, S.A.; Bijani, C.; Canac, Y.; Valyaev, D.A.; Filippov, O.A.; Belkova, N.V.; Shubina, E.S. Fac-to-mer isomerization triggers hydride transfer from Mn (I) complex fac-[(dppm)Mn(CO)3H]. Chem. Comm. 2022, 58, 5017–5020. [Google Scholar] [CrossRef]
- Kaljurand, I.; Kütt, A.; Sooväli, L.; Rodima, T.; Mäemets, V.; Leito, I.; Koppel, I.A. Extension of the self-consistent spectrophotometric basicity scale in acetonitrile to a full span of 28 pKa units: Unification of different basicity scales. J. Org. Chem. 2005, 70, 1019–1028. [Google Scholar] [CrossRef]
- Tabassum, S.; Sereda, O.; Reddy, P.V.G.; Wilhelm, R. Hindered Brønsted bases as Lewis base catalysts. Org. Biomol. Chem. 2009, 7, 4009–4016. [Google Scholar] [CrossRef]
- Narayanan, B.; Amatore, C.; Kochi, J. Electroreduction of carbonylmanganese (I) cations. Mechanism of ligand substitution and hydride formation via manganese (0) intermediates. Organometallics. 1987, 6, 129–136. [Google Scholar] [CrossRef]
- Booth, B.; Haszeldine, R. Metal carbonyl chemistry. Part I. The reactions of phosphorus-containing ligands with hydridopentacarbonylmanganese. J. Chem. Soc. A 1966, 157–160. [Google Scholar] [CrossRef]
- Danopoulos, A.A.; Pugh, D.; Smith, H.; Saßmannshausen, J. Structural and reactivity studies of “pincer” pyridine dicarbene complexes of Fe0: Experimental and computational comparison of the phosphine and NHC donors. Chem. Eur. J. 2009, 15, 5491–5502. [Google Scholar] [CrossRef]
- Zhen, Y.; Feighery, W.G.; Lai, C.K.; Atwood, J.D. Steric and electronic factors that control two-electron processes between metal carbonyl cations and anions. J. Am. Chem. Soc. 1989, 111, 7832–7837. [Google Scholar] [CrossRef]
- Mandal, S.K.; Feldman, J.; Orchin, M. A New route to manganese and rhenium carbonyl tetrafluoroborate salts and an improved procedure for preparing their precursor hydrides. J. Coord. Chem. 1994, 33, 219–221. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.J.A.; Vreven, T.; Kudin, K.N. Gaussian 09, Revision D.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT–Integrated space-group and crystal-structure determination. Acta Crystallogr. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | νCO (cm−1) | δP (ppm) |
|---|---|---|
| 1, [(P(OPh)3)2Mn(CO)3H] | 2040 w, 2028 w, 1956 s a 2041 w, 2028 w, 1955 s b 2043 w, 2031 w, 1958 s c | 183.3 |
| 1+, [(P(OPh)3)2Mn(CO)3][HB(C6F5)3] | 2011 s, 1969 s a | 153.5 |
| 1−, [(P(OPh)3)2Mn(CO)3][HDBU] | 1815 s c | 206.5 |
| 2, [(PPh3)2Mn(CO)3H] | 1908 s, 1900 s f | 80.5 |
| 2+, [(PPh3)2Mn(CO)3]+[HB(C6F5)3]− | 1980 s, 1930 s f | 61.8 |
| 2−, [(PPh3)2Mn(CO)3]−[K+] | 1770 s, 1741 s e | - |
| 3, [(dppm)Mn(CO)3H] | 1996 s, 1916 s, 1909 s a 1993 s, 1914 s, 1903 s b | 30.1 a 31.8 b |
| fac-3+, [(dppm)Mn(CO)3)][HB(C6F5)3] [27] | 2040 s, 1973 s, 1935 s d | 10.1 |
| mer-3+, [(dppm)Mn(CO)3)][HB(C6F5)3] [27] | 2060 s, 2003 s d | 10.6, 7.1 |
| 3CH−, fac–[(CH−-dppm)Mn(CO)3H][K+] | 1957 s, 1871 s, 1876 s e 1956 s, 1870 s b | 10.9 a |
| 3Mn−, [(dppm)Mn−(CO)3][K+] | 1867 s, 1779 s b | 29.9 b |
| 4, fac–[(P–NHC)Mn(CO)3H] | 1989 s, 1909 s, 1889 s a 1988 s, 1907 s,1888 s d | 95.8 |
| fac-4+, [(P–NHC)Mn(CO)3][HB(C6F5)3] | 2032 s, 1949 s, 1921 s d | 78.1, 71.1 f |
| mer-4+, [(P–NHC)Mn(CO)3][HB(C6F5)3] | 2038 s, 1968 s, 1942 s d | - |
| Toluene | THF | MeCN | |
|---|---|---|---|
| Eel | +2.8 | −0.7 | −3.1 |
| ΔH | +5.7 | +2.0 | −0.2 |
| ΔG | +7.6 | +2.5 | −0.2 |
| MnH | keff220K, M−1·s−1 | ∆H≠, kcal/mol | ∆S≠, cal/(mol·K) | ΔG≠220K, kcal/mol | ΔG≠298K, kcal/mol |
|---|---|---|---|---|---|
| 1 | 0.00008 | 8.6 ± 0.2 | −37 ± 1 | 16.9 ± 0.1 | 19.8 ± 0.1 |
| 2 | 0.147 | 4.1 ± 0.5 | −43 ± 3 | 13.6 ± 0.3 | 17.0 ± 0.3 |
| 3 | 0.006 | 9.4 ± 0.6 | −26 ± 3 | 15.0 ± 0.2 | 17.0 ± 0.2 |
| 4 | 0.706 | 3.8 ± 0.2 | −43 ± 1 | 13.2 ± 0.1 | 16.5 ± 0.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osipova, E.S.; Kovalenko, S.A.; Gulyaeva, E.S.; Kireev, N.V.; Pavlov, A.A.; Filippov, O.A.; Danshina, A.A.; Valyaev, D.A.; Canac, Y.; Shubina, E.S.; et al. The Dichotomy of Mn–H Bond Cleavage and Kinetic Hydricity of Tricarbonyl Manganese Hydride Complexes. Molecules 2023, 28, 3368. https://doi.org/10.3390/molecules28083368
Osipova ES, Kovalenko SA, Gulyaeva ES, Kireev NV, Pavlov AA, Filippov OA, Danshina AA, Valyaev DA, Canac Y, Shubina ES, et al. The Dichotomy of Mn–H Bond Cleavage and Kinetic Hydricity of Tricarbonyl Manganese Hydride Complexes. Molecules. 2023; 28(8):3368. https://doi.org/10.3390/molecules28083368
Chicago/Turabian StyleOsipova, Elena S., Sergey A. Kovalenko, Ekaterina S. Gulyaeva, Nikolay V. Kireev, Alexander A. Pavlov, Oleg A. Filippov, Anastasia A. Danshina, Dmitry A. Valyaev, Yves Canac, Elena S. Shubina, and et al. 2023. "The Dichotomy of Mn–H Bond Cleavage and Kinetic Hydricity of Tricarbonyl Manganese Hydride Complexes" Molecules 28, no. 8: 3368. https://doi.org/10.3390/molecules28083368
APA StyleOsipova, E. S., Kovalenko, S. A., Gulyaeva, E. S., Kireev, N. V., Pavlov, A. A., Filippov, O. A., Danshina, A. A., Valyaev, D. A., Canac, Y., Shubina, E. S., & Belkova, N. V. (2023). The Dichotomy of Mn–H Bond Cleavage and Kinetic Hydricity of Tricarbonyl Manganese Hydride Complexes. Molecules, 28(8), 3368. https://doi.org/10.3390/molecules28083368