

Studying the Mechanism of Interaction of Doxofylline with Human Lysozyme: A Biophysical and In Silico Approach


Abstract
:1. Introduction
2. Results and Discussion
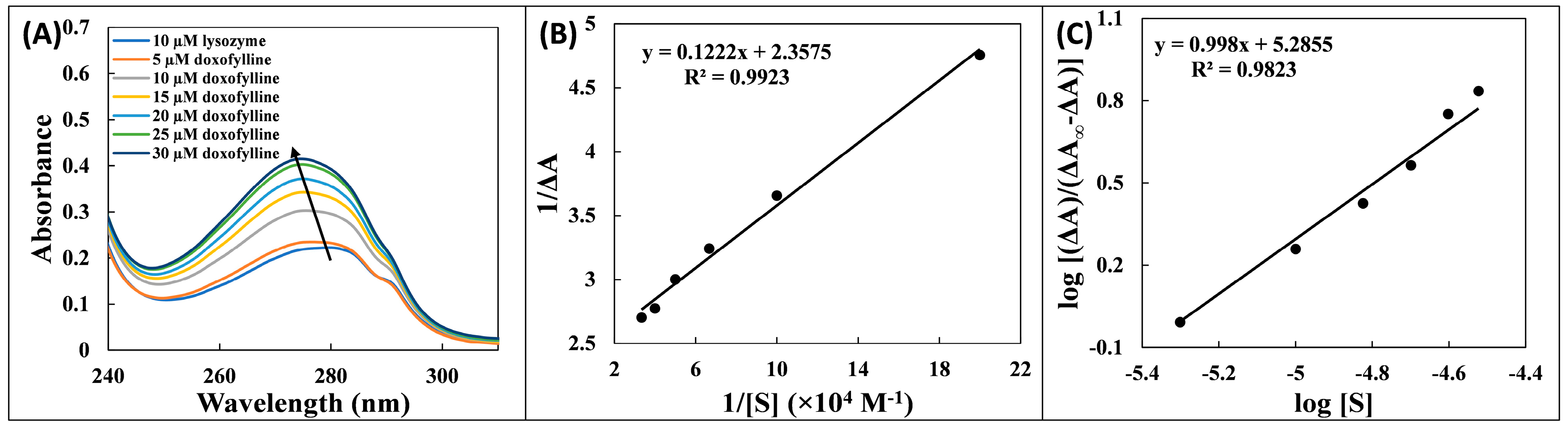
2.1. UV Absorption Spectroscopy Study
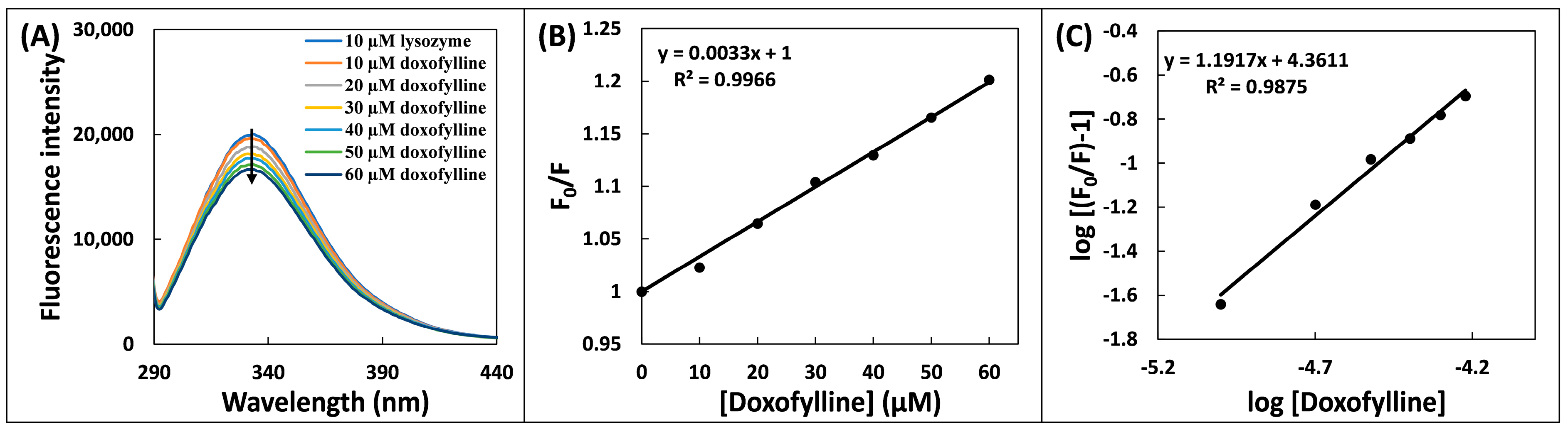
2.2. Steady-State Fluorescence Spectroscopic Analysis
2.3. Synchronous Fluorescence Examination
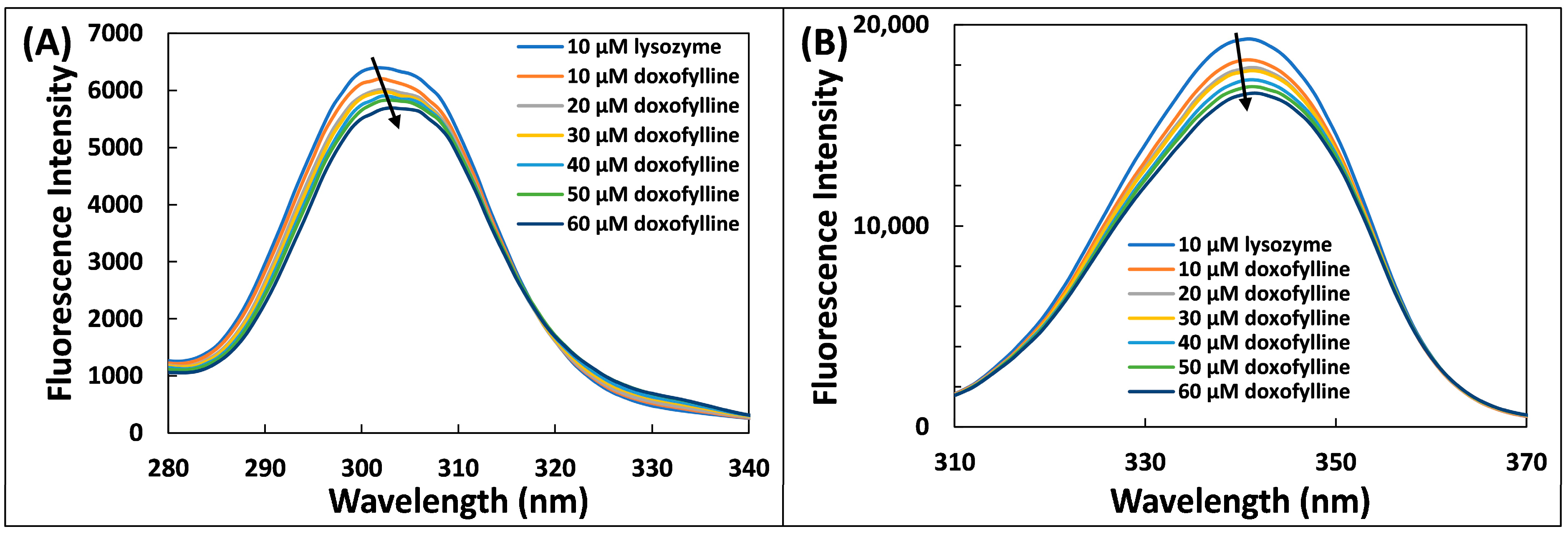
2.4. Three Dimensional (3D)-Fluorescence Spectroscopic Study
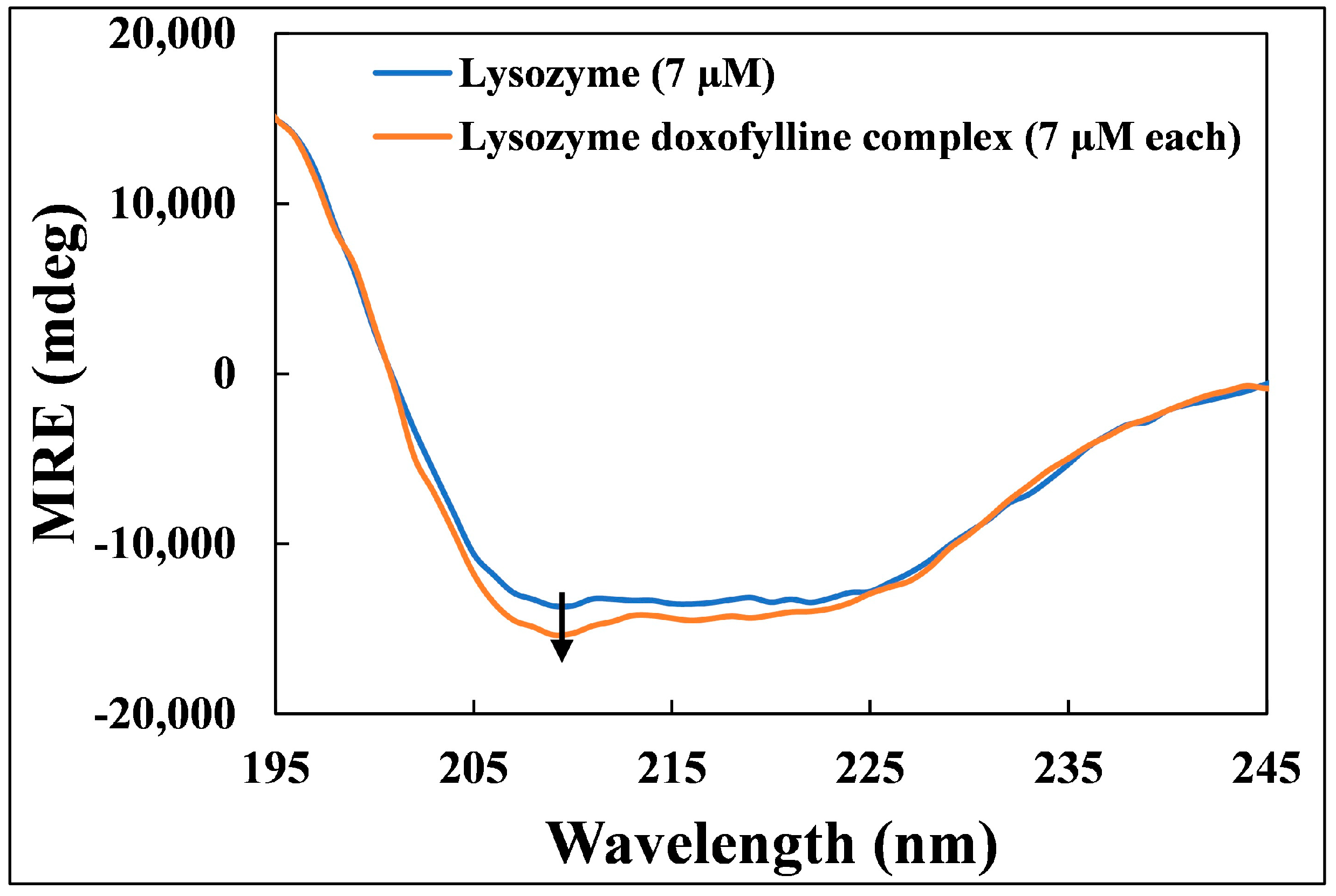
2.5. Circular Dichroism (CD) Measurements
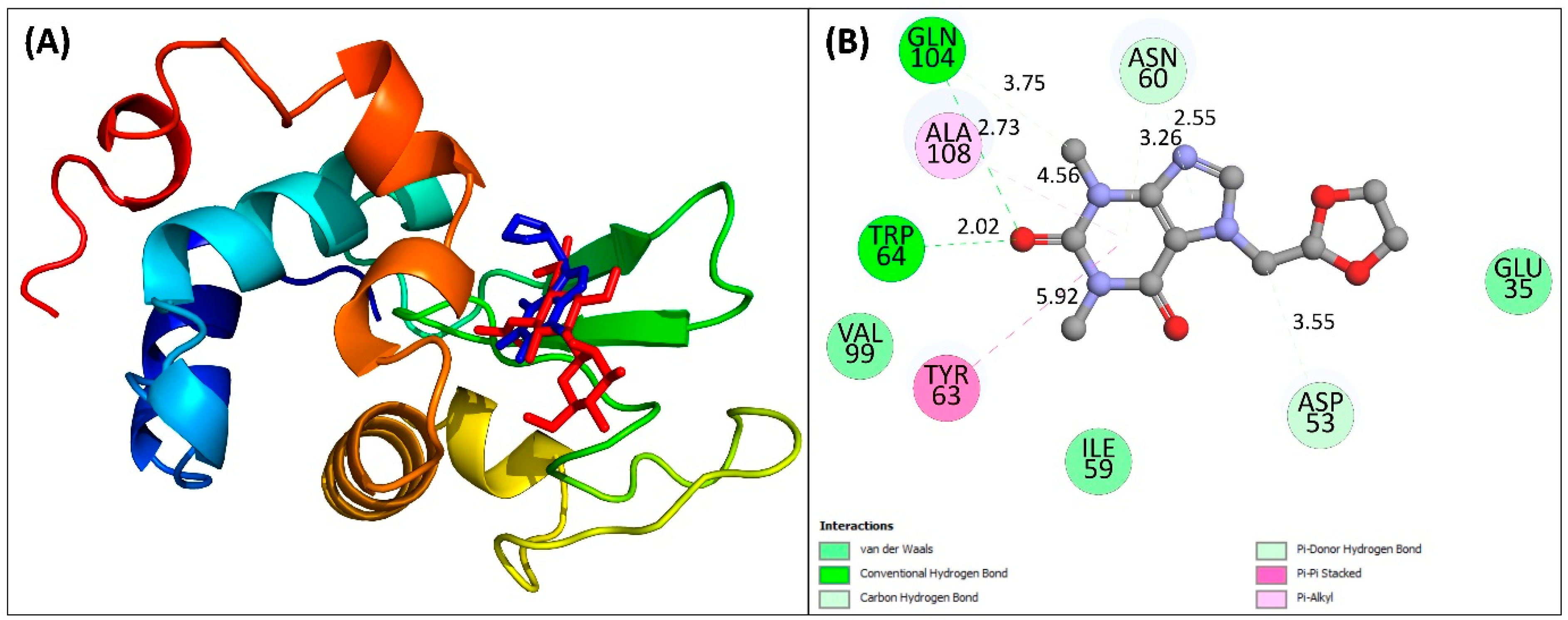
2.6. Molecular Docking Study
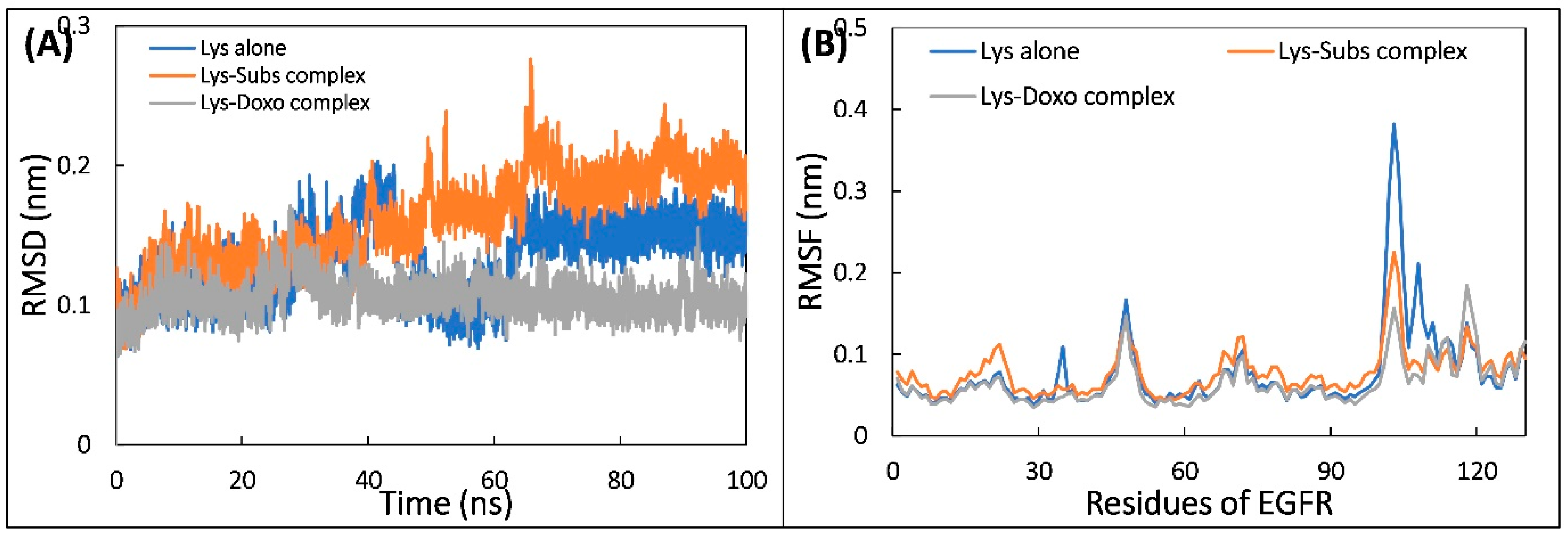
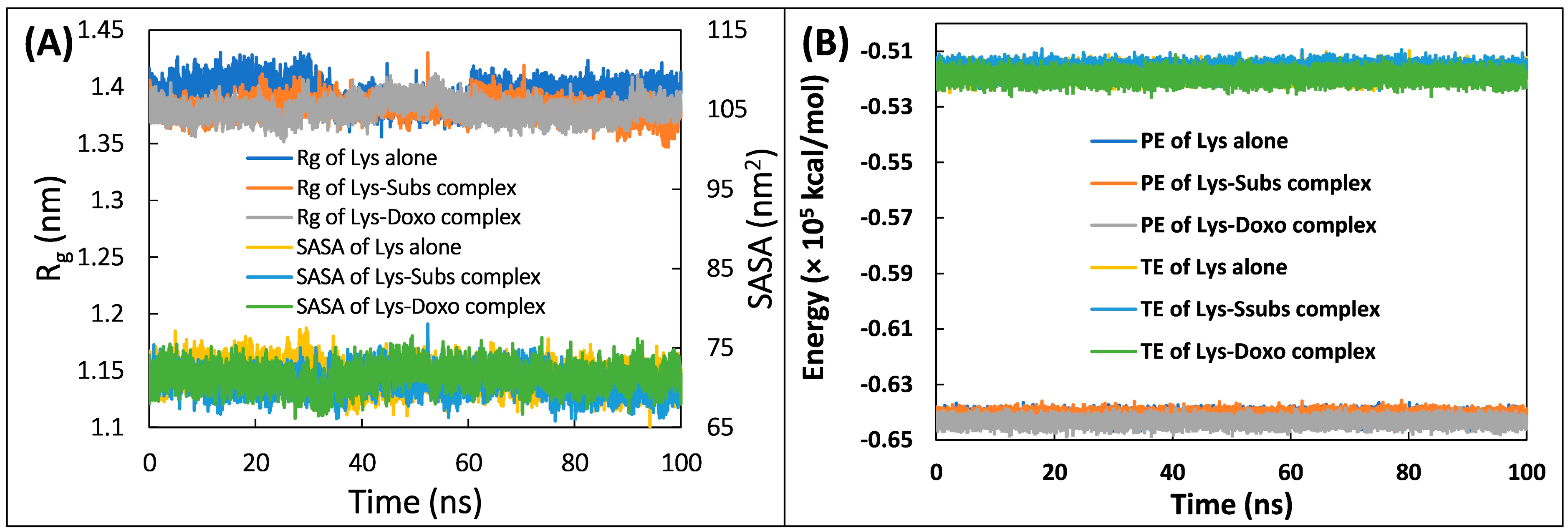
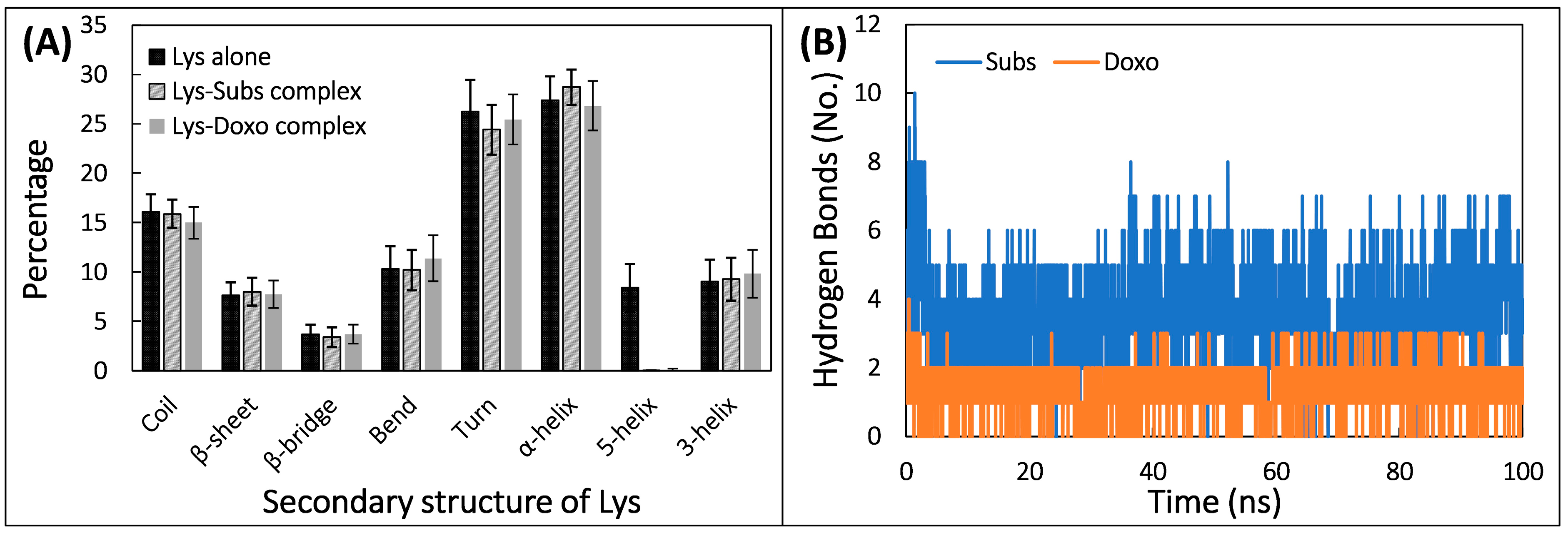
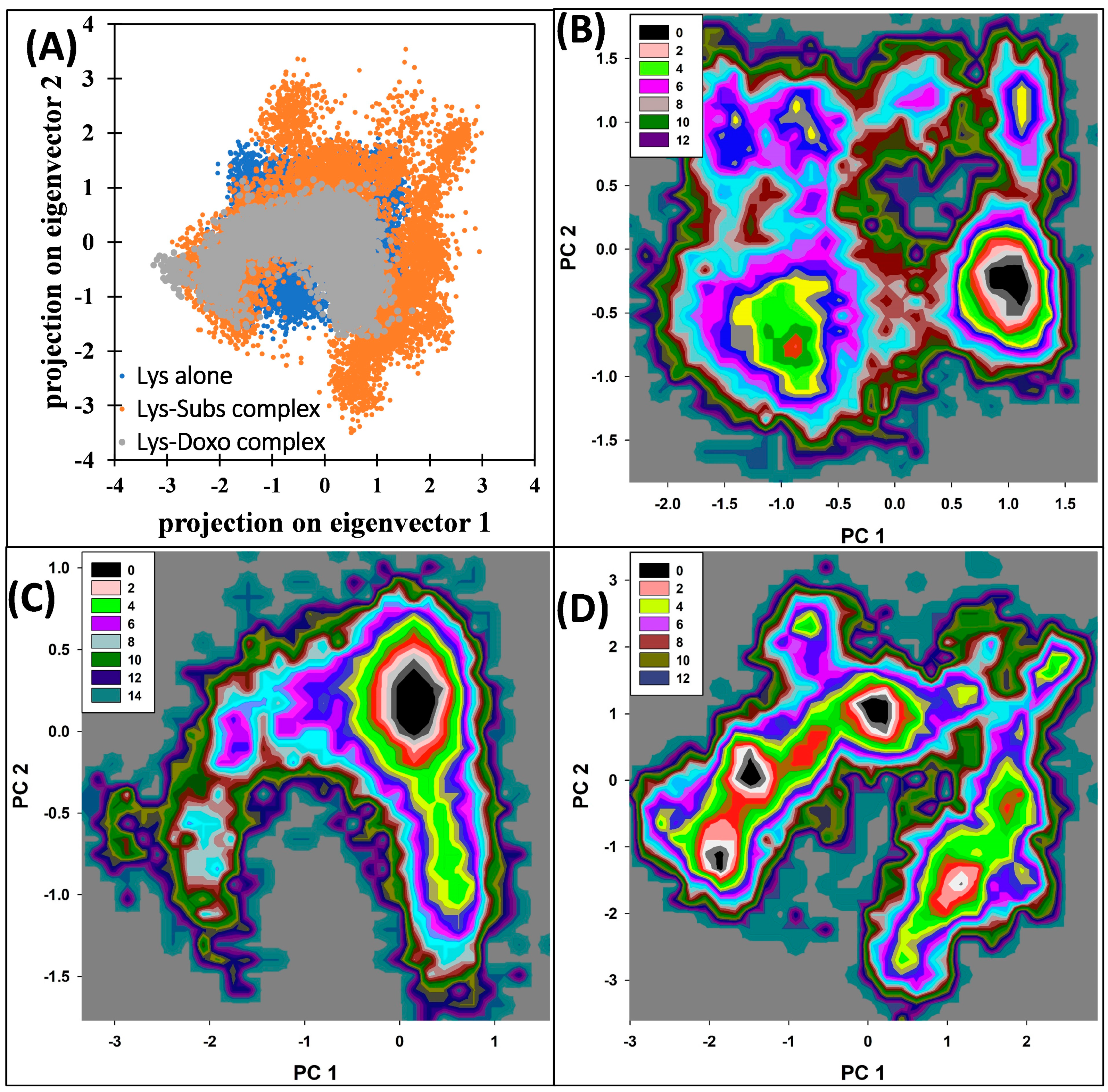
2.7. Molecular Dynamic Simulation
3. Experimental Materials and Methods
3.1. Materials
3.2. Sample Preparation
3.3. Methods
3.3.1. UV Absorption Spectroscopy Study
3.3.2. Fluorescence Quenching Experiment
3.3.3. Synchronous Fluorescence Examination
3.3.4. 3D (Three Dimensional) Fluorescence Emission Spectroscopic Study
3.3.5. Circular Dichroism (CD) Measurements
3.3.6. Molecular Docking
3.3.7. Molecular Dynamic Simulation
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ferraboschi, P.; Ciceri, S.; Grisenti, P. Applications of Lysozyme, an Innate Immune Defense Factor, as an Alternative Antibiotic. Antibiotics 2021, 10, 1534. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.M.; Barik, S.; Preeyanka, N.; Sarkar, M. Interaction of Lysozyme with Monocationic and Dicationic Ionic Liquids: Toward Finding a Suitable Medium for Biomacromolecules. J. Phys. Chem. B 2020, 124, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Oliver, W.T.; Wells, J.E. Lysozyme as an Alternative to Growth Promoting Antibiotics in Swine Production. J. Anim. Sci. Biotechnol. 2015, 6, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiorentini, R.; Kremer, K.; Potestio, R. Ligand-protein Interactions in Lysozyme Investigated through a Dual-resolution Model. Proteins Struct. Funct. Bioinform. 2020, 88, 1351–1360. [Google Scholar] [CrossRef]
- Asemi-Esfahani, Z.; Shareghi, B.; Farhadian, S.; Momeni, L. Effect of Naphthol Yellow S as a Food Dye on the Lysozyme Structure and Its Mechanisms of Action. J. Mol. Liq. 2021, 332, 115846. [Google Scholar] [CrossRef]
- Silvetti, T.; Morandi, S.; Hintersteiner, M.; Brasca, M. Use of Hen Egg White Lysozyme in the Food Industry. In Egg Innovations and Strategies for Improvements; Elsevier: Amsterdam, The Netherlands, 2017; pp. 233–242. [Google Scholar]
- Hughey, V.L.; Johnson, E.A. Antimicrobial Activity of Lysozyme against Bacteria Involved in Food Spoilage and Food-Borne Disease. Appl. Environ. Microbiol. 1987, 53, 2165–2170. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Hazarika, Z.; Sarmah, S.; Baruah, K.; Rohman, M.A.; Paul, D.; Jha, A.N.; Singha Roy, A. Exploring the Interaction of Bioactive Kaempferol with Serum Albumin, Lysozyme and Hemoglobin: A Biophysical Investigation Using Multi-Spectroscopic, Docking and Molecular Dynamics Simulation Studies. J. Photochem. Photobiol. B Biol. 2020, 205, 111825. [Google Scholar] [CrossRef]
- Millan, S.; Satish, L.; Bera, K.; Susrisweta, B.; Singh, D.V.; Sahoo, H. A Spectroscopic and Molecular Simulation Approach toward the Binding Affinity between Lysozyme and Phenazinium Dyes: An Effect on Protein Conformation. J. Phys. Chem. B 2017, 121, 1475–1484. [Google Scholar] [CrossRef]
- Jash, C.; Payghan, P.V.; Ghoshal, N.; Suresh Kumar, G. Binding of the Iminium and Alkanolamine Forms of Sanguinarine to Lysozyme: Spectroscopic Analysis, Thermodynamics, and Molecular Modeling Studies. J. Phys. Chem. B 2014, 118, 13077–13091. [Google Scholar] [CrossRef]
- Das, S.; Pahari, S.; Sarmah, S.; Rohman, M.A.; Paul, D.; Jana, M.; Singha Roy, A. Lysozyme–Luteolin Binding: Molecular Insights into the Complexation Process and the Inhibitory Effects of Luteolin towards Protein Modification. Phys. Chem. Chem. Phys. 2019, 21, 12649–12666. [Google Scholar] [CrossRef]
- Page, C.P. Doxofylline: A “Novofylline”. Pulm. Pharmacol. Ther. 2010, 23, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Matera, M.G.; Page, C.; Cazzola, M. Doxofylline Is Not Just Another Theophylline! Int. J. Chronic Obstruct. Pulmon. Dis. 2017, 12, 3487–3493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haj Ahmed, W.; Peiro, C.; Fontaine, J.; Ryan, B.J.; Kinsella, G.K.; O’Sullivan, J.; Grolleau, J.-L.; Henehan, G.T.M.; Carpéné, C. Methylxanthines Inhibit Primary Amine Oxidase and Monoamine Oxidase Activities of Human Adipose Tissue. Medicines 2020, 7, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, M.; Song, W.; Liu, R. Binding of Copper to Lysozyme: Spectroscopic, Isothermal Titration Calorimetry and Molecular Docking Studies. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2016, 164, 103–109. [Google Scholar] [CrossRef]
- Ahmad, E.; Rabbani, G.; Zaidi, N.; Singh, S.; Rehan, M.; Khan, M.M.; Rahman, S.K.; Quadri, Z.; Shadab, M.; Ashraf, M.T.; et al. Stereo-Selectivity of Human Serum Albumin to Enantiomeric and Isoelectronic Pollutants Dissected by Spectroscopy, Calorimetry and Bioinformatics. PLoS ONE 2011, 6, e26186. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, S.; Ameen, F.; Jahan, I.; Nayeem, S.M.; Tabish, M. A Comprehensive Spectroscopic and Computational Investigation on the Binding of the Anti-Asthmatic Drug Triamcinolone with Serum Albumin. New J. Chem. 2019, 43, 4137–4151. [Google Scholar] [CrossRef]
- Pawar, S.; Raul, K.; Ottoor, D. Investigation of Complexation of Amlodipine with Lysozyme and Its Effect on Lysozyme Crystal Growth. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 227, 117623. [Google Scholar] [CrossRef]
- Khan, S.N.; Islam, B.; Yennamalli, R.; Sultan, A.; Subbarao, N.; Khan, A.U. Interaction of Mitoxantrone with Human Serum Albumin: Spectroscopic and Molecular Modeling Studies. Eur. J. Pharm. Sci. 2008, 35, 371–382. [Google Scholar] [CrossRef]
- González-Durruthy, M.; Rial, R.; Liu, Z.; Ruso, J.M. Lysozyme Allosteric Interactions with β-Blocker Drugs. J. Mol. Liq. 2022, 366, 120370. [Google Scholar] [CrossRef]
- Qais, F.A.; Alam, M.M.; Naseem, I.; Ahmad, I. Understanding the Mechanism of Non-Enzymatic Glycation Inhibition by Cinnamic Acid: An in Vitro Interaction and Molecular Modelling Study. RSC Adv. 2016, 6, 65322–65337. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alam, M.M.; Qais, F.A.; Ahmad, I.; Alam, P.; Hasan Khan, R.; Naseem, I. Multi-Spectroscopic and Molecular Modelling Approach to Investigate the Interaction of Riboflavin with Human Serum Albumin. J. Biomol. Struct. Dyn. 2018, 36, 795–809. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Hao, C.; Liu, H.; Yang, H.; Zhong, K.; Zhang, M.; Sun, R. Interaction of Graphene Oxide with Lysozyme:Insights from Conformational Structure and Surface Charge Investigations. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2022, 264, 120207. [Google Scholar] [CrossRef]
- Siddiqui, S.; Mujeeb, A.; Ameen, F.; Ishqi, H.M.; Rehman, S.U.; Tabish, M. Investigating the Mechanism of Binding of Nalidixic Acid with Deoxyribonucleic Acid and Serum Albumin: A Biophysical and Molecular Docking Approaches. J. Biomol. Struct. Dyn. 2021, 39, 570–585. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.; Weljie, A.M.; Vogel, H.J. Tryptophan Fluorescence Quenching by Methionine and Selenomethionine Residues of Calmodulin: Orientation of Peptide and Protein Binding. Biochemistry 1998, 37, 3187–3195. [Google Scholar] [CrossRef]
- Kragh-Hansen, U. Molecular Aspects of Ligand Binding to Serum Albumin. Pharmacol. Rev. 1981, 33, 17–53. [Google Scholar]
- Han, R.; Liu, B.; Li, G.; Zhang, Q. Investigation on the Interaction of Cefpirome Sulfate with Lysozyme by Fluorescence Quenching Spectroscopy and Synchronous Fluorescence Spectroscopy. Luminescence 2016, 31, 580–586. [Google Scholar] [CrossRef]
- Głowacz, K.; Skorupska, S.; Grabowska-Jadach, I.; Ciosek-Skibińska, P. Excitation–Emission Matrix Fluorescence Spectroscopy for Cell Viability Testing in UV-Treated Cell Culture. RSC Adv. 2022, 12, 7652–7660. [Google Scholar] [CrossRef]
- Lyndem, S.; Gazi, R.; Jana, M.; Belwal, V.K.; Singha Roy, A. Molecular Recognition of Two Bioactive Coumarin Derivatives 7-Hydroxycoumarin and 4-Methyl-7-Hydroxycoumarin by Hen Egg White Lysozyme: Exploring the Binding Mechanism, Thermodynamic Parameters and Structural Changes Using Multispectroscopic and Computati. J. Biomol. Struct. Dyn. 2021, 40, 13872–13888. [Google Scholar] [CrossRef]
- Khatun, S.; Riyazuddeen; Qais, F.A. Characterization of the Binding of Triprolidine Hydrochloride to Hen Egg White Lysozyme by Multi-Spectroscopic and Molecular Docking Techniques. J. Mol. Liq. 2018, 269, 521–528. [Google Scholar] [CrossRef]
- Moradi, S.; Shareghi, B.; Saboury, A.A.; Farhadian, S. Investigation on the Interaction of Acid Phosphatase with Putrescine Using Docking, Simulations Methods and Multispectroscopic Techniques. Int. J. Biol. Macromol. 2020, 150, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Riley, K.; Tran, K.-A. Strength and Character of R–X···π Interactions Involving Aromatic Amino Acid Sidechains in Protein-Ligand Complexes Derived from Crystal Structures in the Protein Data Bank. Crystals 2017, 7, 273. [Google Scholar] [CrossRef] [Green Version]
- Ouassaf, M.; Belaidi, S.; Chtita, S.; Lanez, T.; Abul Qais, F.; Md Amiruddin, H. Combined Molecular Docking and Dynamics Simulations Studies of Natural Compounds as Potent Inhibitors against SARS-CoV-2 Main Protease. J. Biomol. Struct. Dyn. 2021, 40, 11264–11273. [Google Scholar] [CrossRef] [PubMed]
- Nour, H.; Abchir, O.; Belaidi, S.; Qais, F.A.; Chtita, S.; Belaaouad, S. 2D-QSAR and Molecular Docking Studies of Carbamate Derivatives to Discover Novel Potent Anti-butyrylcholinesterase Agents for Alzheimer’s Disease Treatment. Bull. Korean Chem. Soc. 2022, 43, 277–292. [Google Scholar] [CrossRef]
- Belhassan, A.; Zaki, H.; Chtita, S.; Alaqarbeh, M.; Alsakhen, N.; Benlyas, M.; Lakhlifi, T.; Bouachrine, M. Camphor, Artemisinin and Sumac Phytochemicals as Inhibitors against COVID-19: Computational Approach. Comput. Biol. Med. 2021, 136, 104758. [Google Scholar] [CrossRef]
- Qais, F.A.; Sarwar, T.; Ahmad, I.; Khan, R.A.; Shahzad, S.A.; Husain, F.M. Glyburide Inhibits Non-Enzymatic Glycation of HSA: An Approach for the Management of AGEs Associated Diabetic Complications. Int. J. Biol. Macromol. 2021, 169, 143–152. [Google Scholar] [CrossRef]
- Rath, B.; Abul Qais, F.; Patro, R.; Mohapatra, S.; Sharma, T. Design, Synthesis and Molecular Modeling Studies of Novel Mesalamine Linked Coumarin for Treatment of Inflammatory Bowel Disease. Bioorg. Med. Chem. Lett. 2021, 41, 128029. [Google Scholar] [CrossRef]
- Belhassan, A.; Chtita, S.; Zaki, H.; Alaqarbeh, M.; Alsakhen, N.; Almohtaseb, F.; Lakhlifi, T.; Bouachrine, M. In Silico Detection of Potential Inhibitors from Vitamins and Their Derivatives Compounds against SARS-CoV-2 Main Protease by Using Molecular Docking, Molecular Dynamic Simulation and ADMET Profiling. J. Mol. Struct. 2022, 1258, 132652. [Google Scholar] [CrossRef]
- Fouedjou, R.T.; Chtita, S.; Bakhouch, M.; Belaidi, S.; Ouassaf, M.; Djoumbissie, L.A.; Tapondjou, L.A.; Abul Qais, F. Cameroonian Medicinal Plants as Potential Candidates of SARS-CoV-2 Inhibitors. J. Biomol. Struct. Dyn. 2021, 40, 8615–8629. [Google Scholar] [CrossRef]
- Jafari, M.; Mehrnejad, F. Molecular Insight into Human Lysozyme and Its Ability to Form Amyloid Fibrils in High Concentrations of Sodium Dodecyl Sulfate: A View from Molecular Dynamics Simulations. PLoS ONE 2016, 11, e0165213. [Google Scholar] [CrossRef] [Green Version]
- Siddiqui, S.; Ameen, F.; Kausar, T.; Nayeem, S.M.; Ur Rehman, S.; Tabish, M. Biophysical Insight into the Binding Mechanism of Doxofylline to Bovine Serum Albumin: An in Vitro and in Silico Approach. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 249, 119296. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhang, Q.; Wang, F.; Yuan, L.; Xu, Z.; Jiang, F.; Liu, Y. Comparison of Interactions between Human Serum Albumin and Silver Nanoparticles of Different Sizes Using Spectroscopic Methods. Luminescence 2014, 30, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warren, L.; DeLano, P.D. PyMOL: An Open-Source Molecular Graphics Tool. CCP4 Newsl. Protein Cryst. 2002, 40, 82–92. [Google Scholar]
- BIOVIA, Dassault Systèmes. BIOVIA Workbook, Release 2020; BIOVIA Pipeline Pilot, Release 2020; Dassault Systèmes: San Diego, CA, USA, 2023. [Google Scholar]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. Ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from Ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [Green Version]
- Sousa da Silva, A.W.; Vranken, W.F. ACPYPE—AnteChamber PYthon Parser InterfacE. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. G_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Temperature | Kd (M) | Ka (M−1) | h | ΔG0 (kcal mol−1) |
|---|---|---|---|---|
| 298 K | 5.18 × 10−6 M | 19.29 × 104 M−1 | 0.998 | −7.20 |
| Lysozyme:Doxofylline | Peak 1 280/340 | Peak 2 230/340 | Peak a 280/280 | Peak b 280/540 |
|---|---|---|---|---|
| 1:0 | 96,680.4 | 35,574.9 | 244,663.4 | 5036.2 |
| 1:2 | 96,008.5 | 34,751.4 | 229,976.1 | 4752.9 |
| Lysozyme Conc. | Doxofylline Conc. | Lysozyme:Doxofylline | MRE208 nm | % α-Helix |
|---|---|---|---|---|
| 7 | 0 | 1:0 | −13,273.95 | 31.97 |
| 7 | 7 | 1:1 | −14,891.64 | 37.55 |
| Energy Type | Ligand | |
|---|---|---|
| Substrate | Doxofylline | |
| ΔEvdW | −51.99 ± 0.33 | −42.22 ± 0.37 |
| ΔEele | −38.60 ± 0.28 | −61.84 ± 1.23 |
| ΔEPSE | 72.70 ± 0.48 | 78.62 ± 1.24 |
| ΔESSASA | −5.25 ± 0.01 | −5.12 ± 0.01 |
| ΔEBE | −23.11 ± 0.47 | −30.55 ± 0.56 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alomar, S.Y. Studying the Mechanism of Interaction of Doxofylline with Human Lysozyme: A Biophysical and In Silico Approach. Molecules 2023, 28, 3462. https://doi.org/10.3390/molecules28083462
Alomar SY. Studying the Mechanism of Interaction of Doxofylline with Human Lysozyme: A Biophysical and In Silico Approach. Molecules. 2023; 28(8):3462. https://doi.org/10.3390/molecules28083462
Chicago/Turabian StyleAlomar, Suliman Yousef. 2023. "Studying the Mechanism of Interaction of Doxofylline with Human Lysozyme: A Biophysical and In Silico Approach" Molecules 28, no. 8: 3462. https://doi.org/10.3390/molecules28083462
APA StyleAlomar, S. Y. (2023). Studying the Mechanism of Interaction of Doxofylline with Human Lysozyme: A Biophysical and In Silico Approach. Molecules, 28(8), 3462. https://doi.org/10.3390/molecules28083462