
New Approved Drugs Appearing in the Pharmaceutical Market in 2022 Featuring Fragments of Tailor-Made Amino Acids and Fluorine

 ,
,  , and
, and 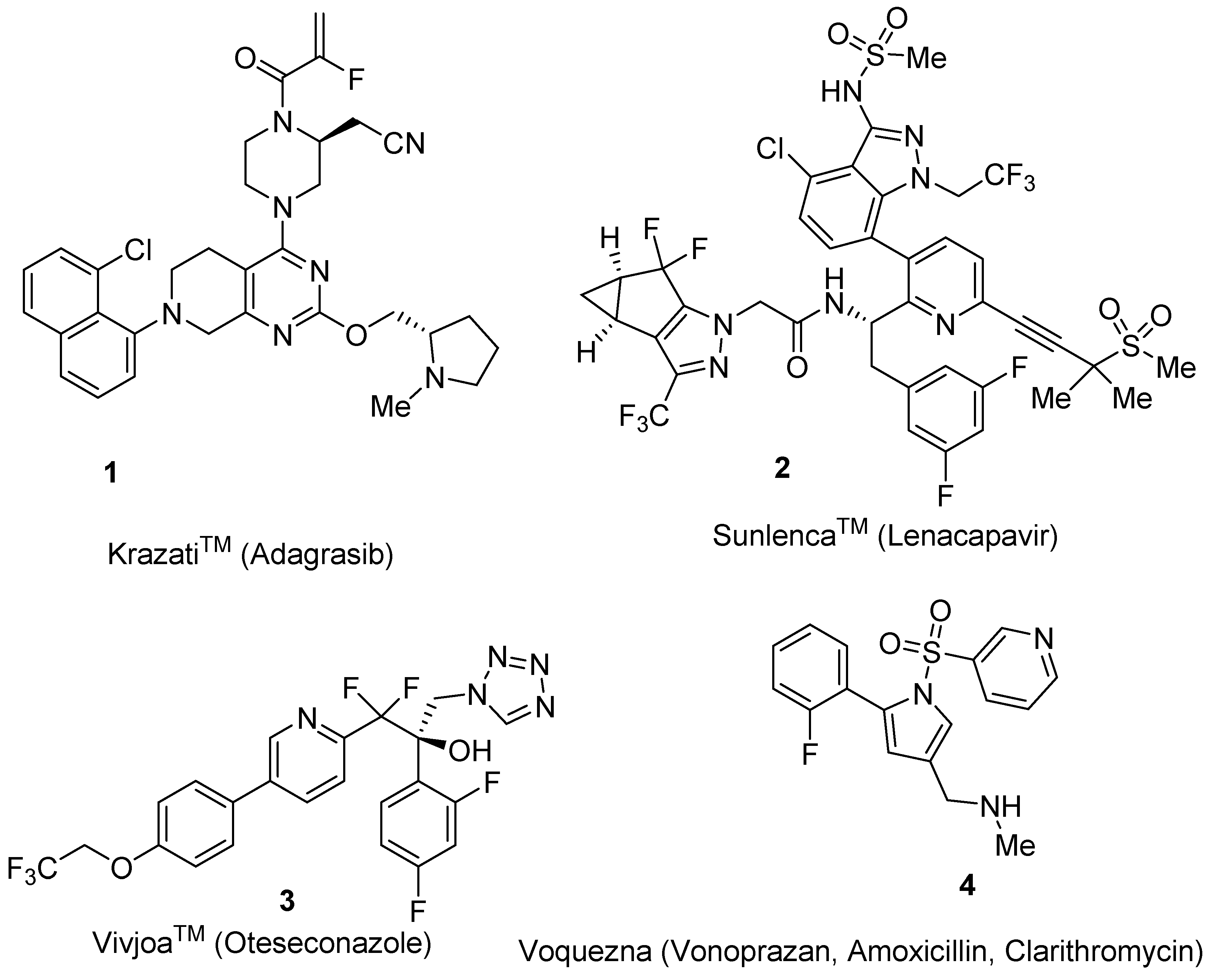{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Fluorine-Containing Drugs
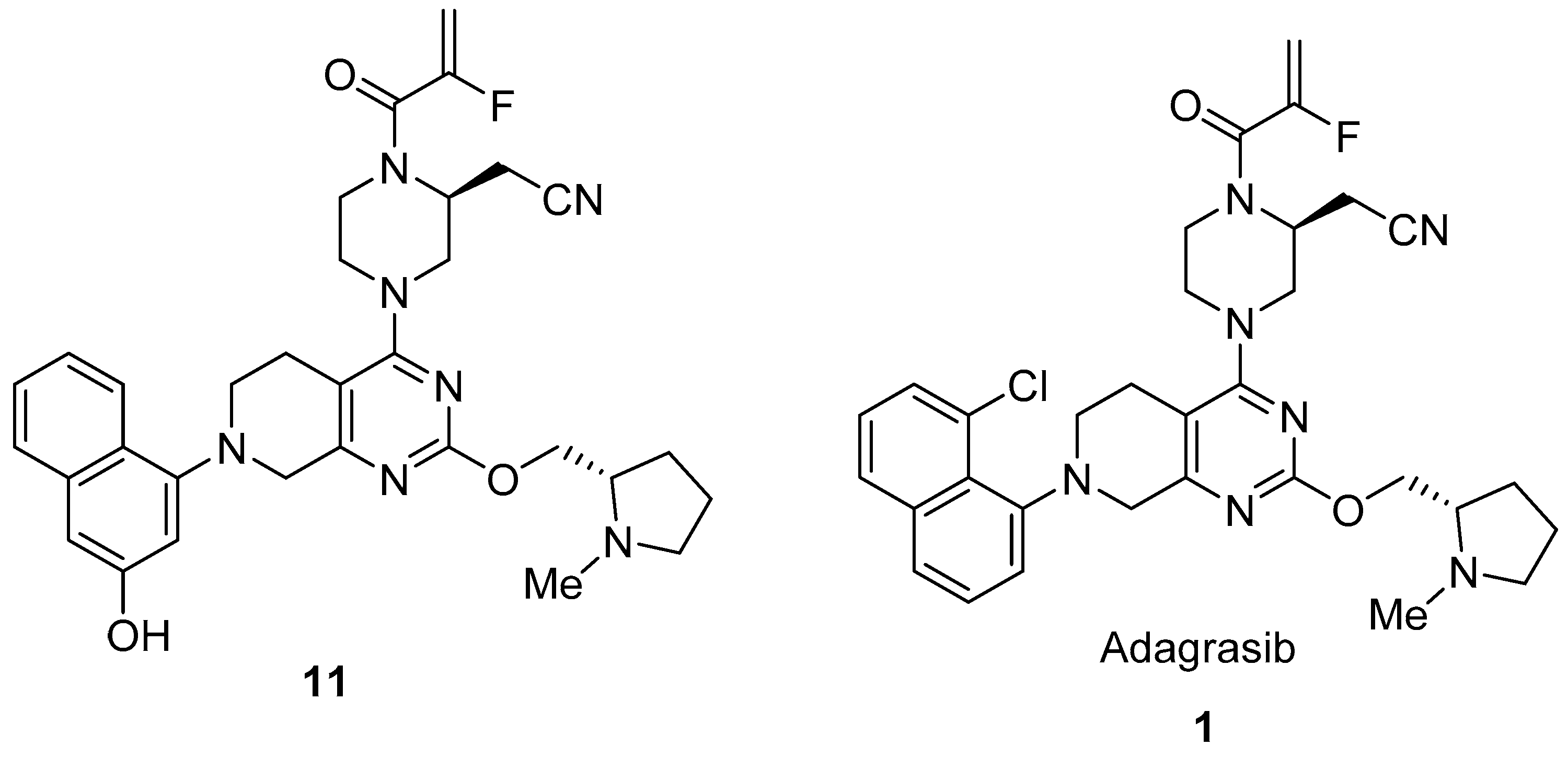
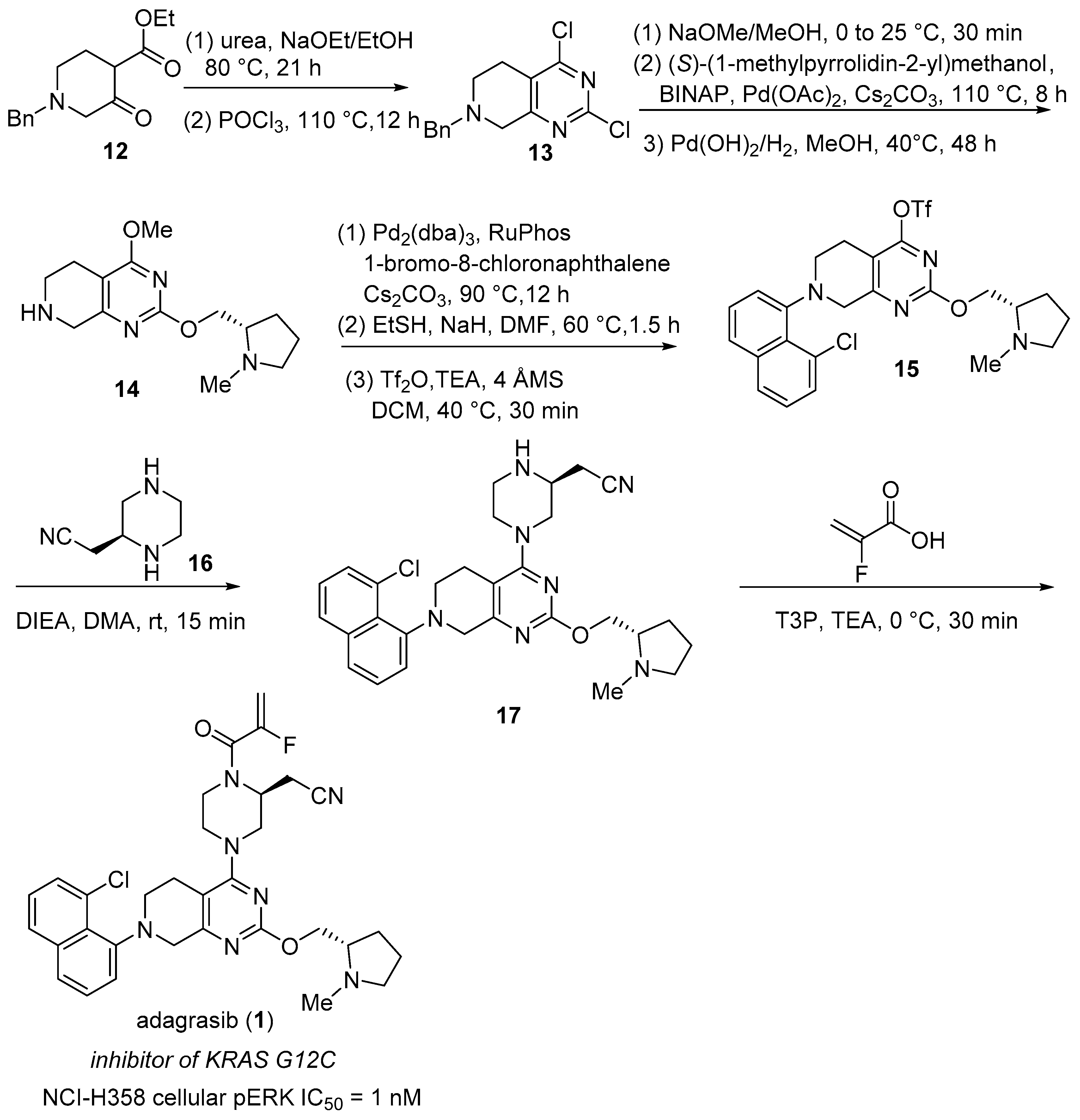
2.1. Adagrasib (KrazatiTM)
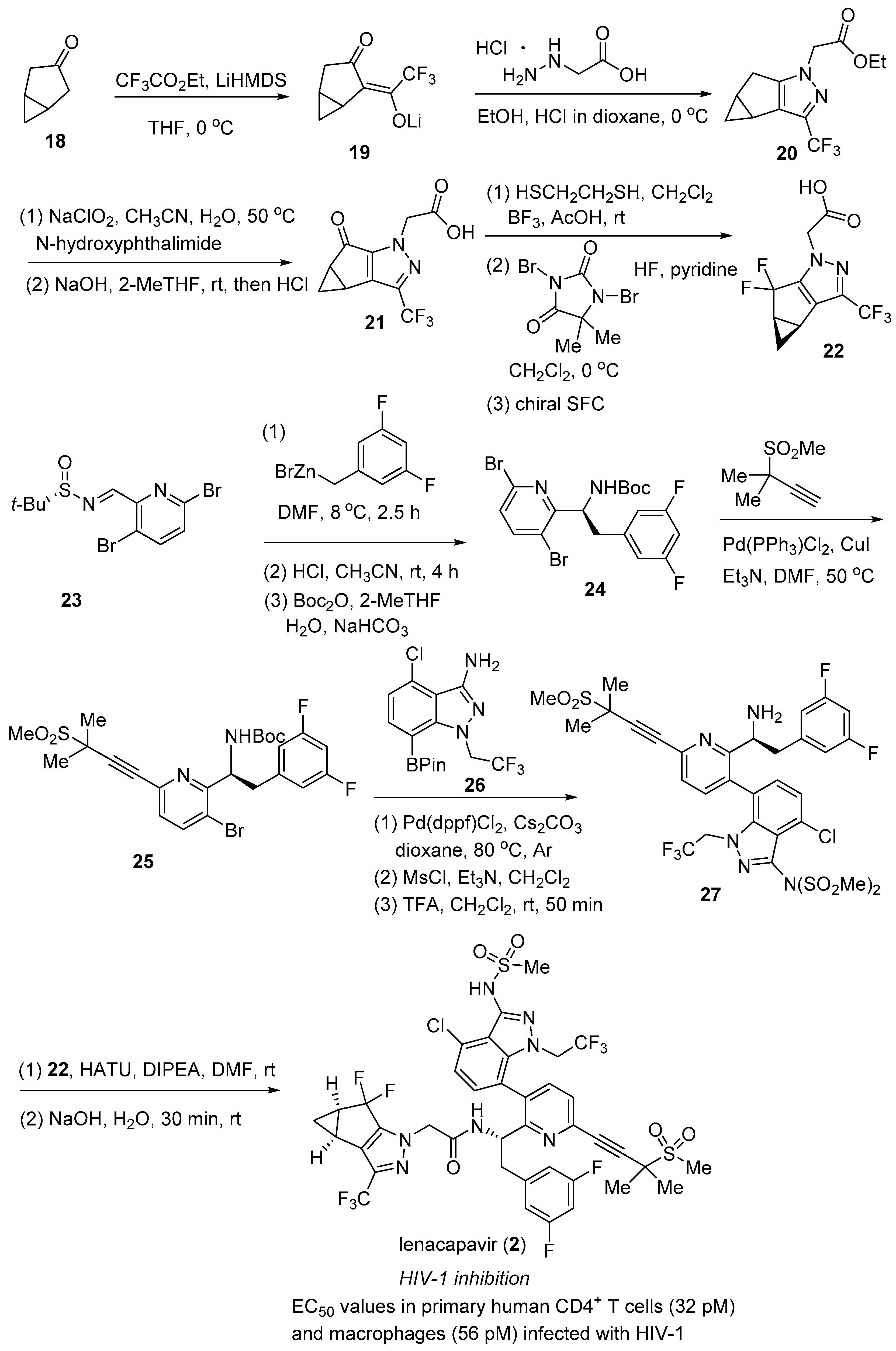
2.2. Lenacapavir (SunlencaTM)
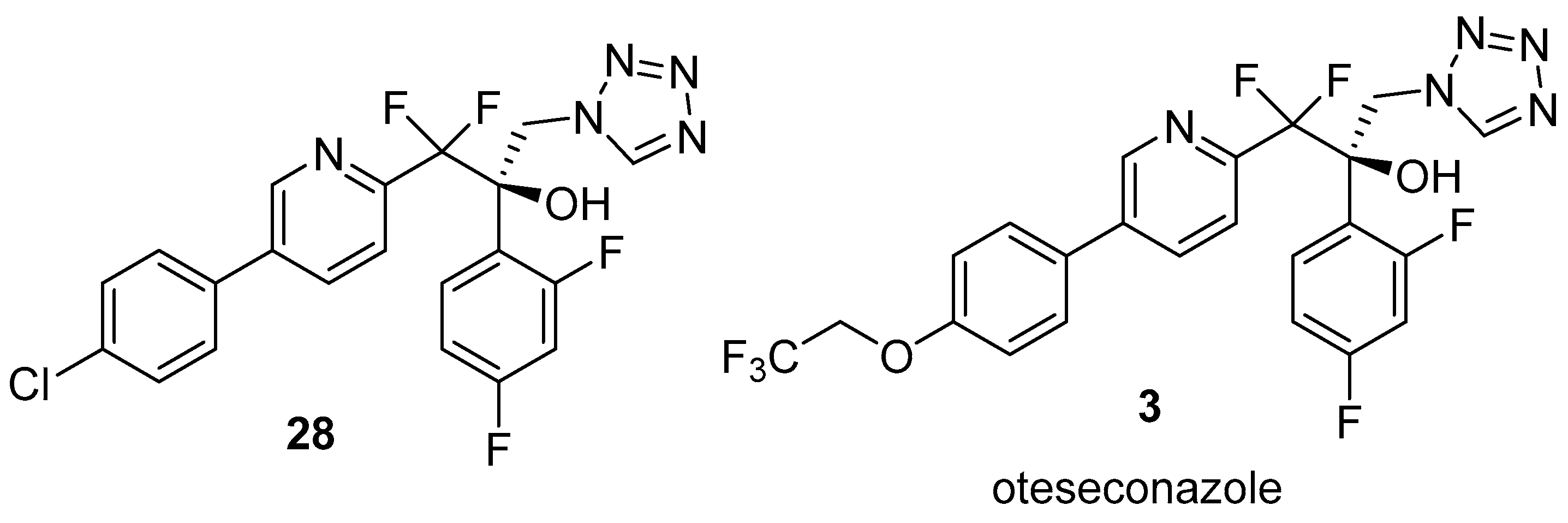
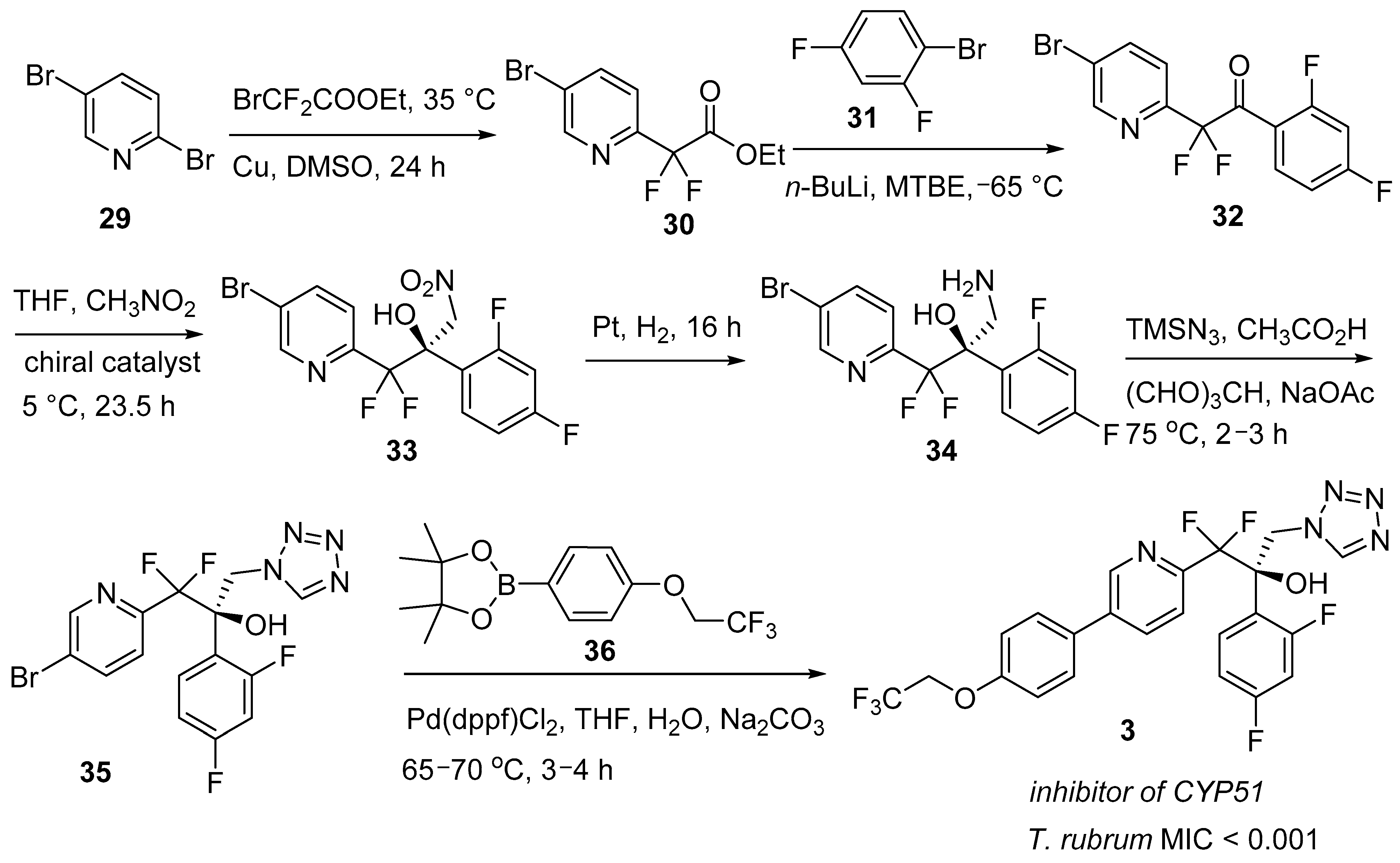
2.3. Oteseconazole (Vivjoa™)
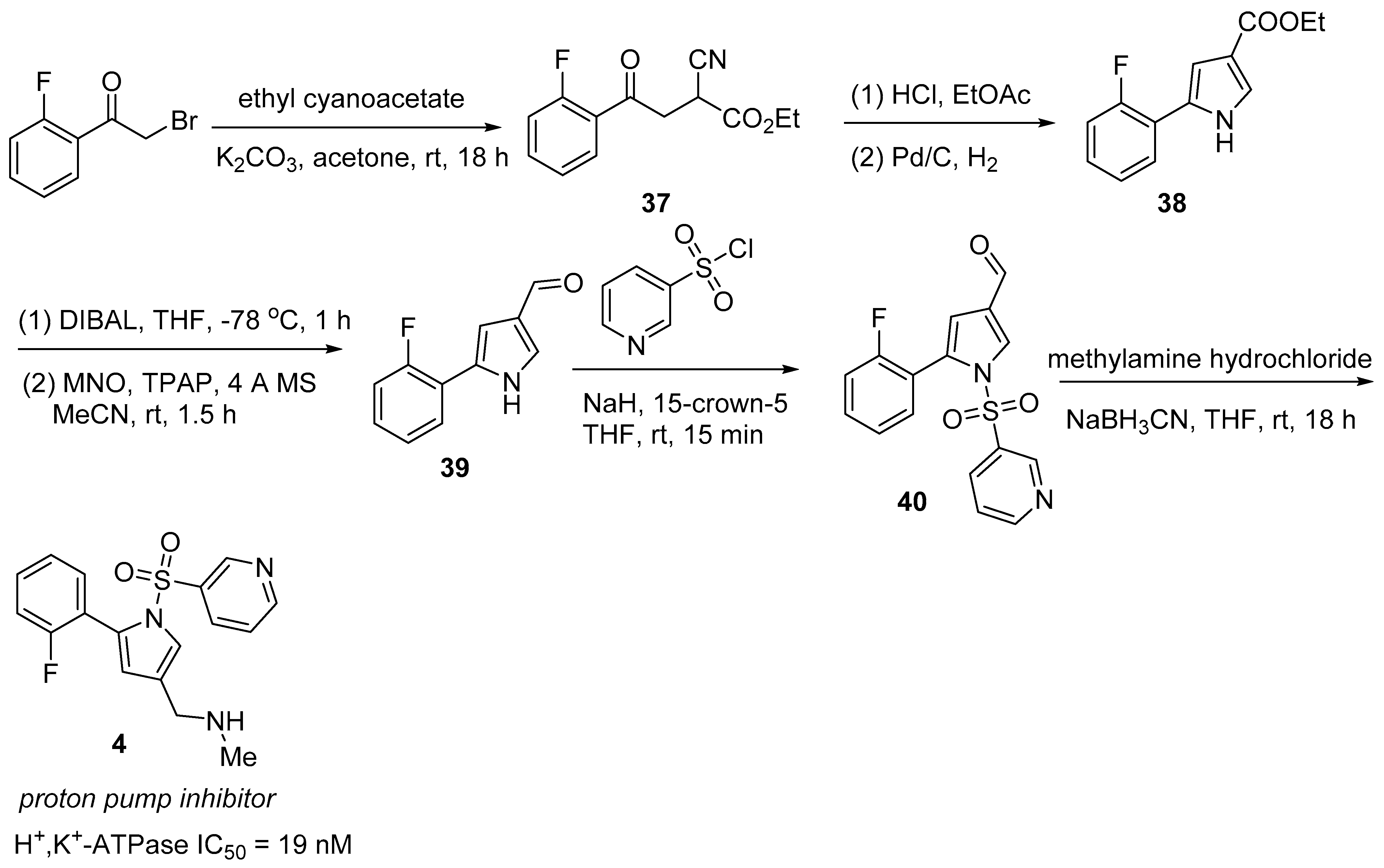
2.4. Vonoprazan/Amoxicillin/Clarithromycin (VoqueznaTM)
3. AA-Derived Drugs
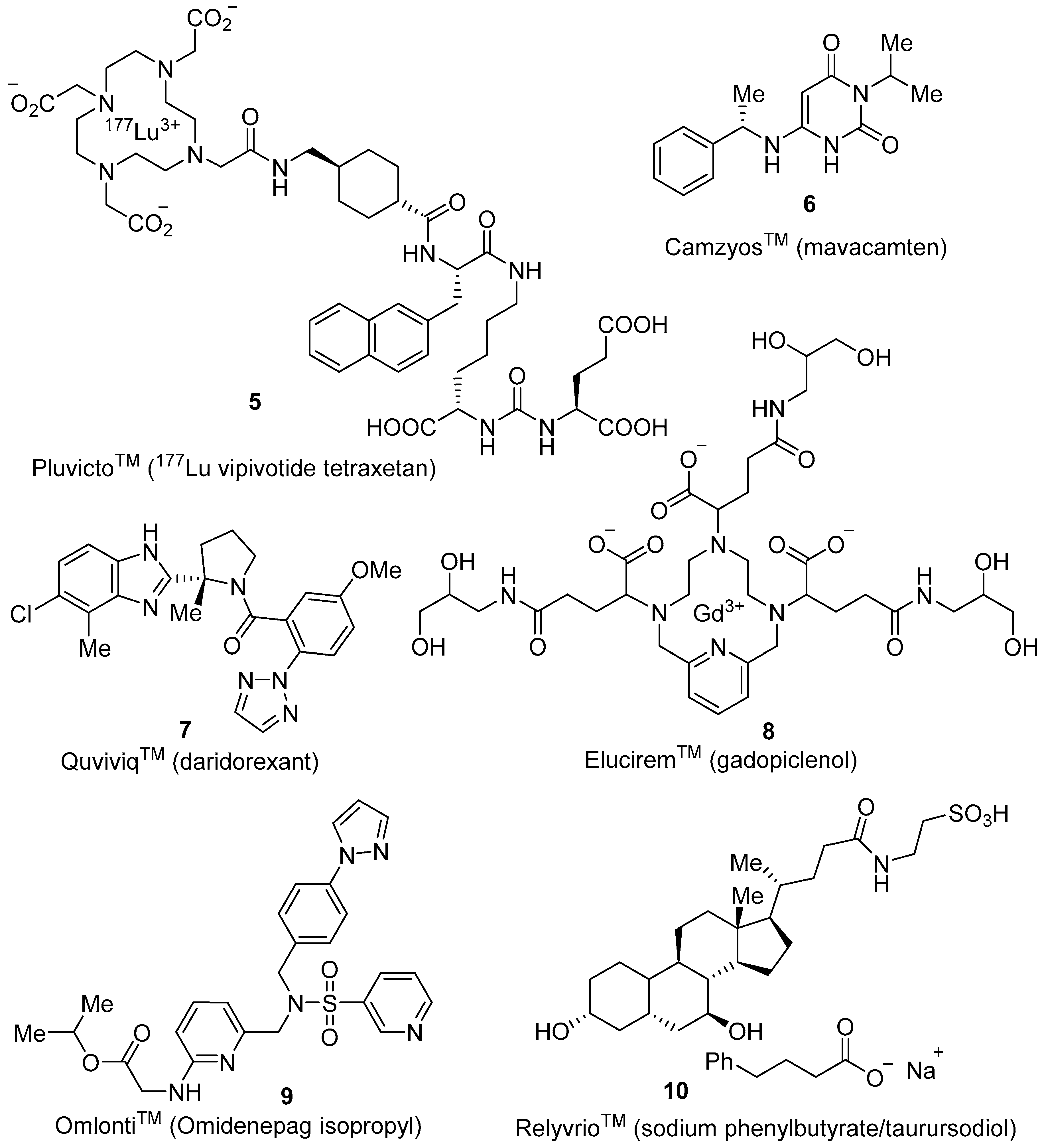
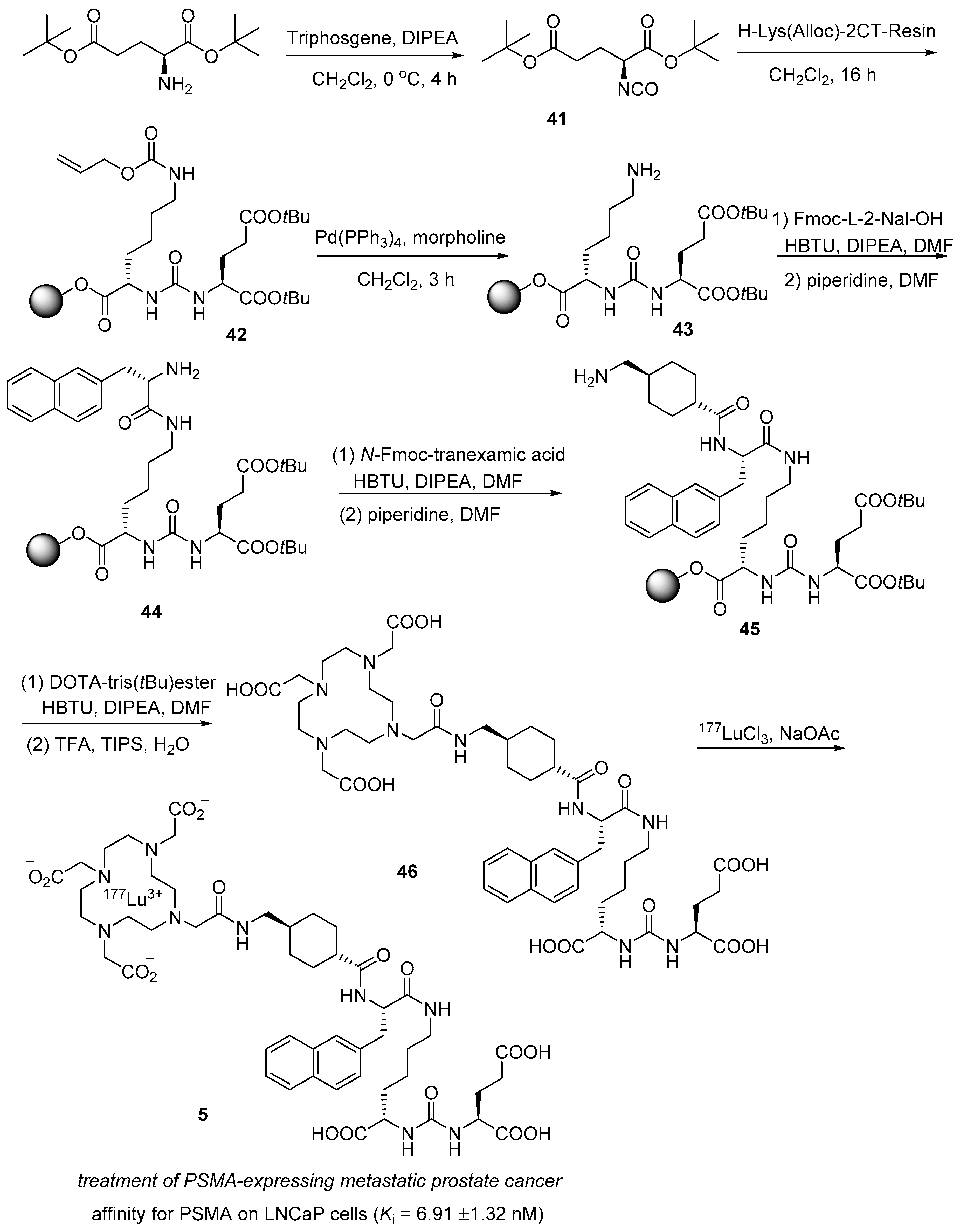
3.1. 177Lu Vipivotide Tetraxetan (PluvictoTM)
3.2. Mavacamten (CamzyosTM)
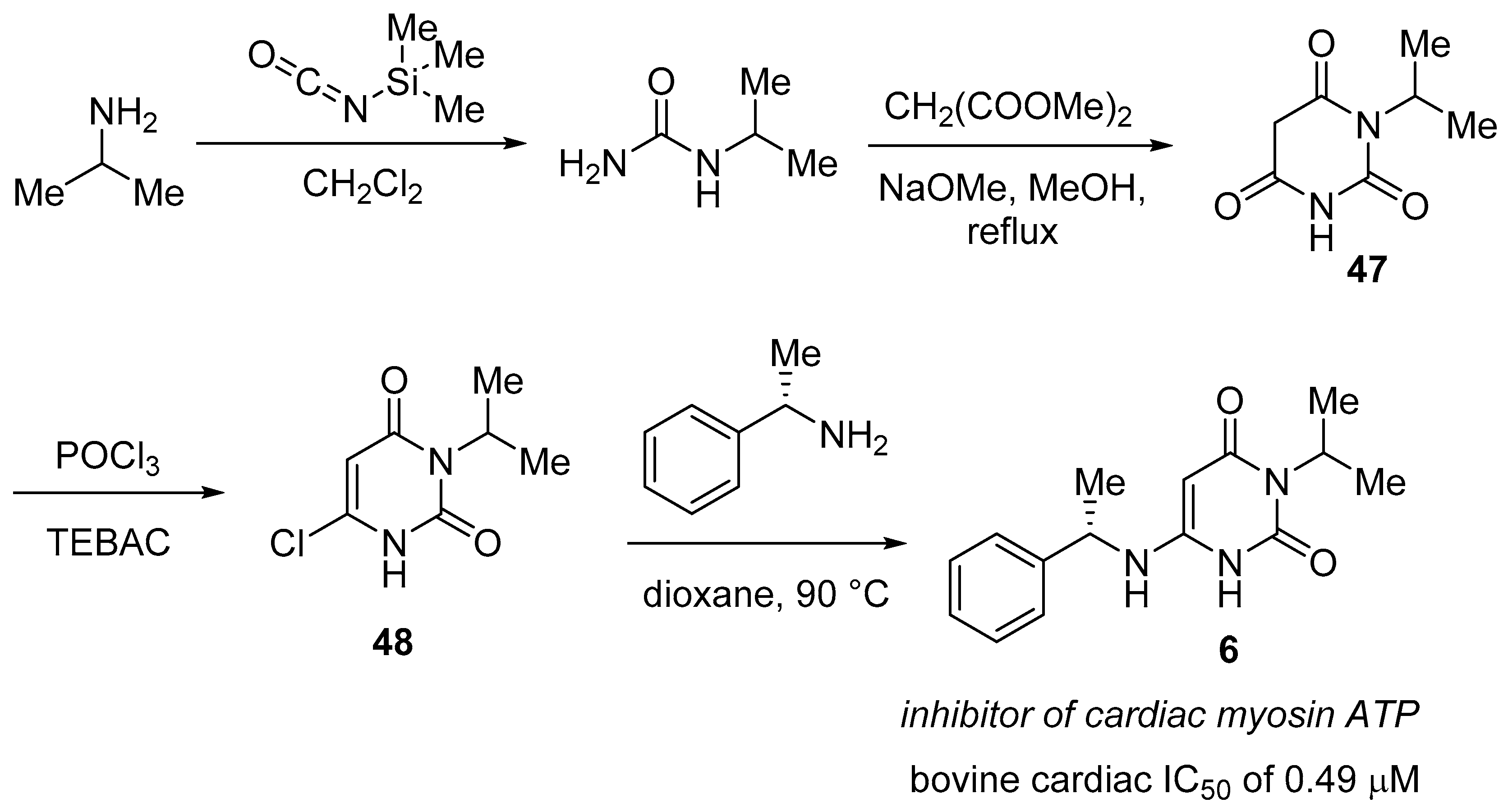
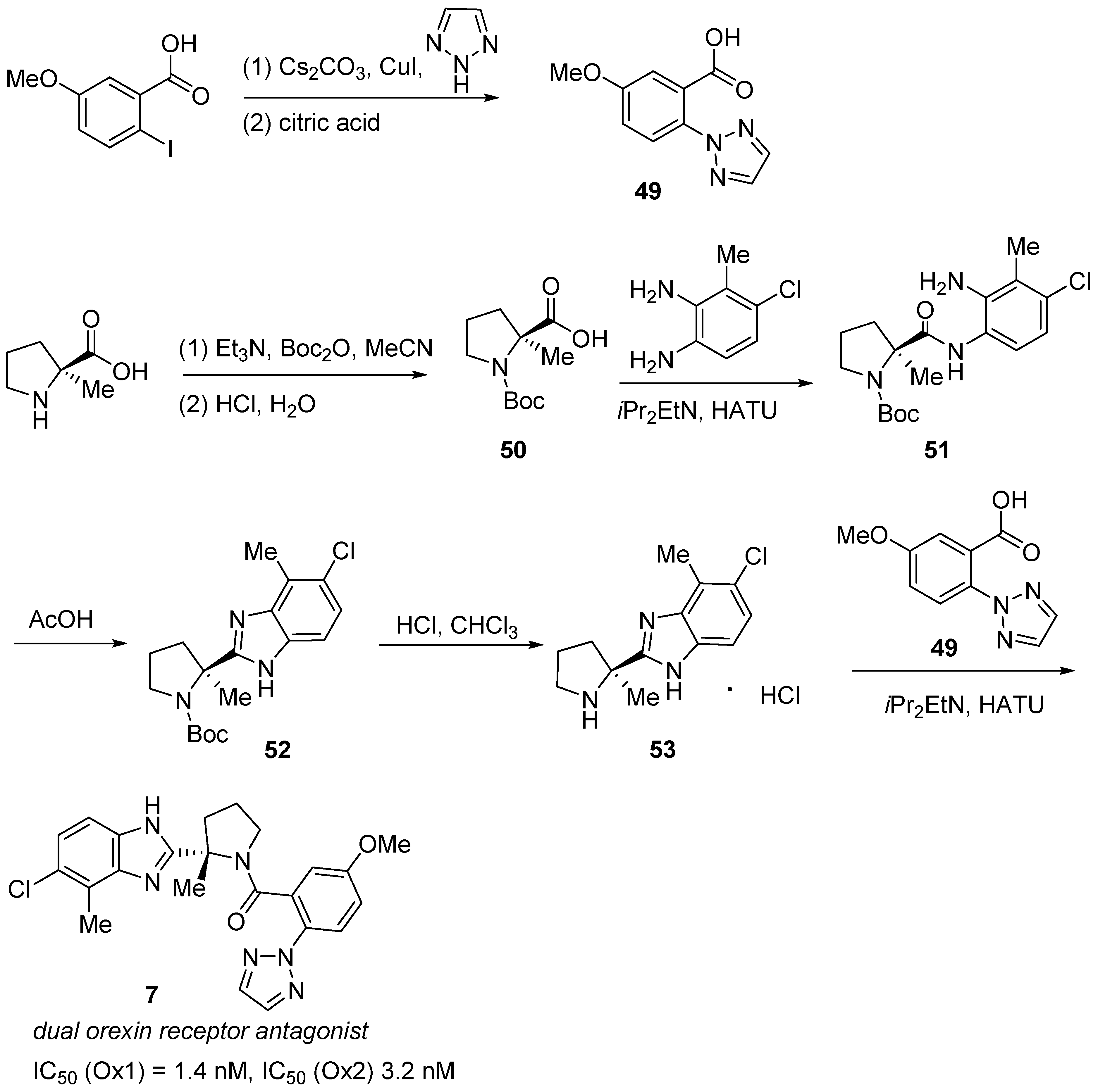
3.3. Daridorexant (QuviviqTM)
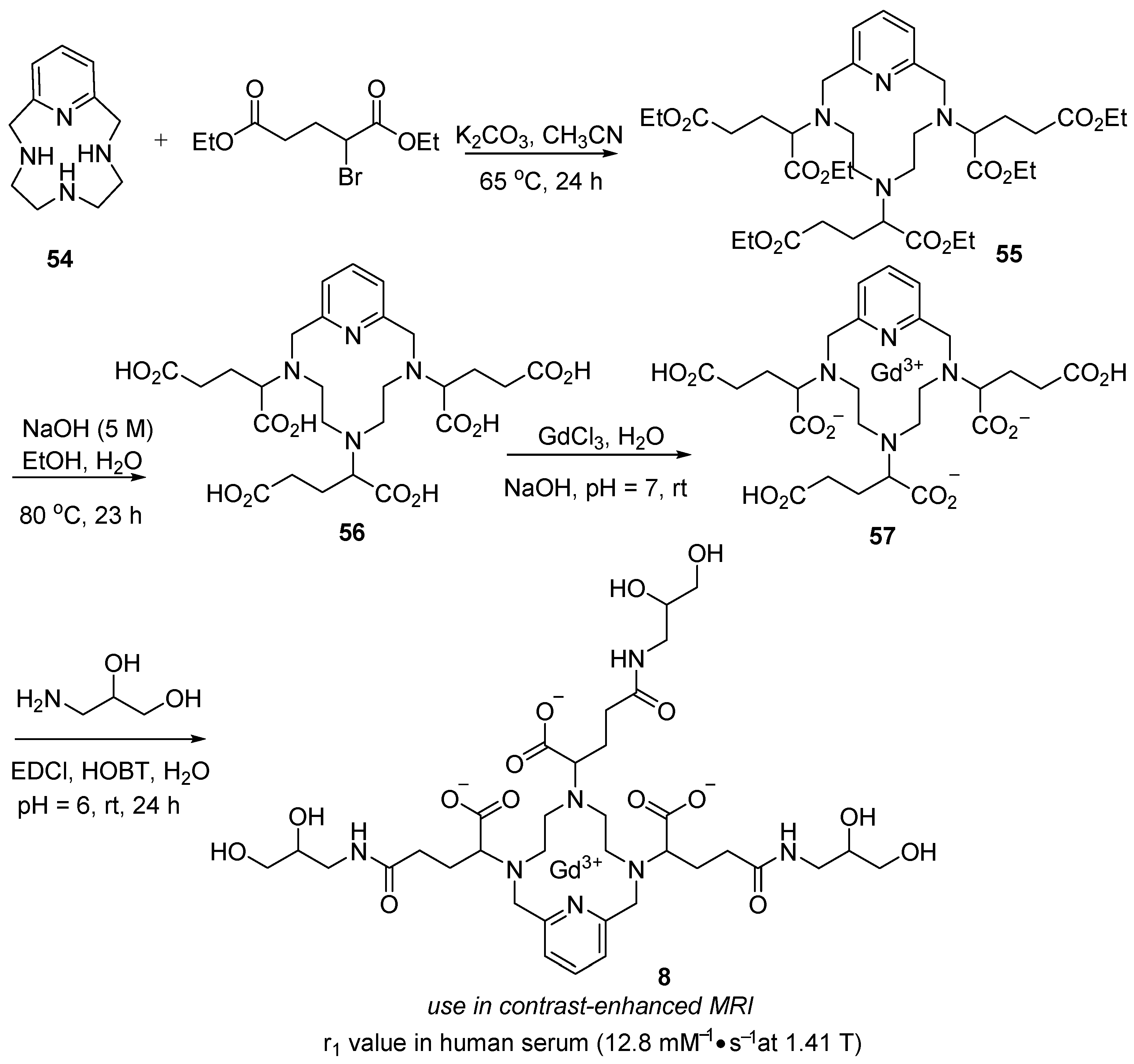
3.4. Gadopiclenol (EluciremTM)
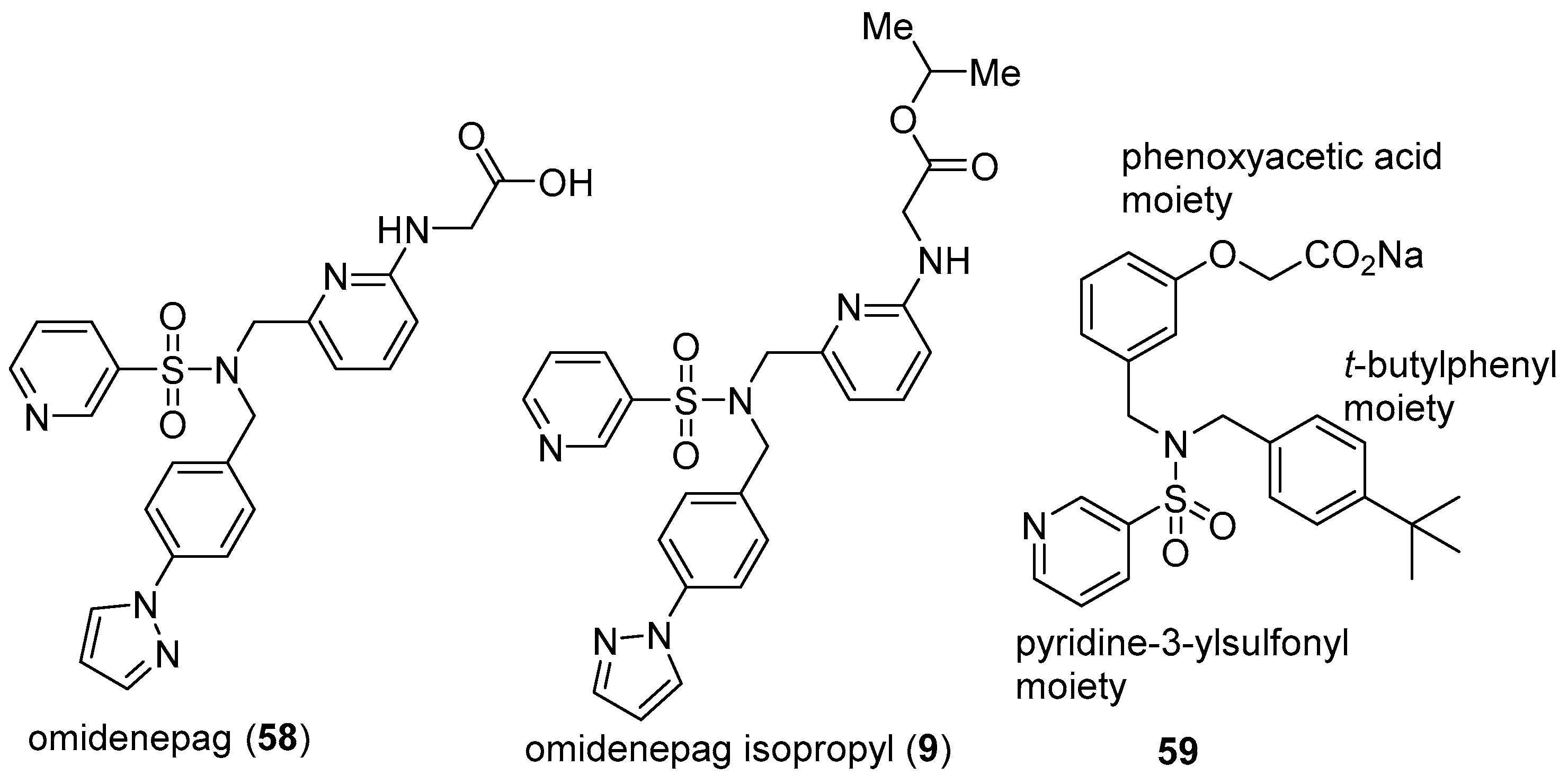
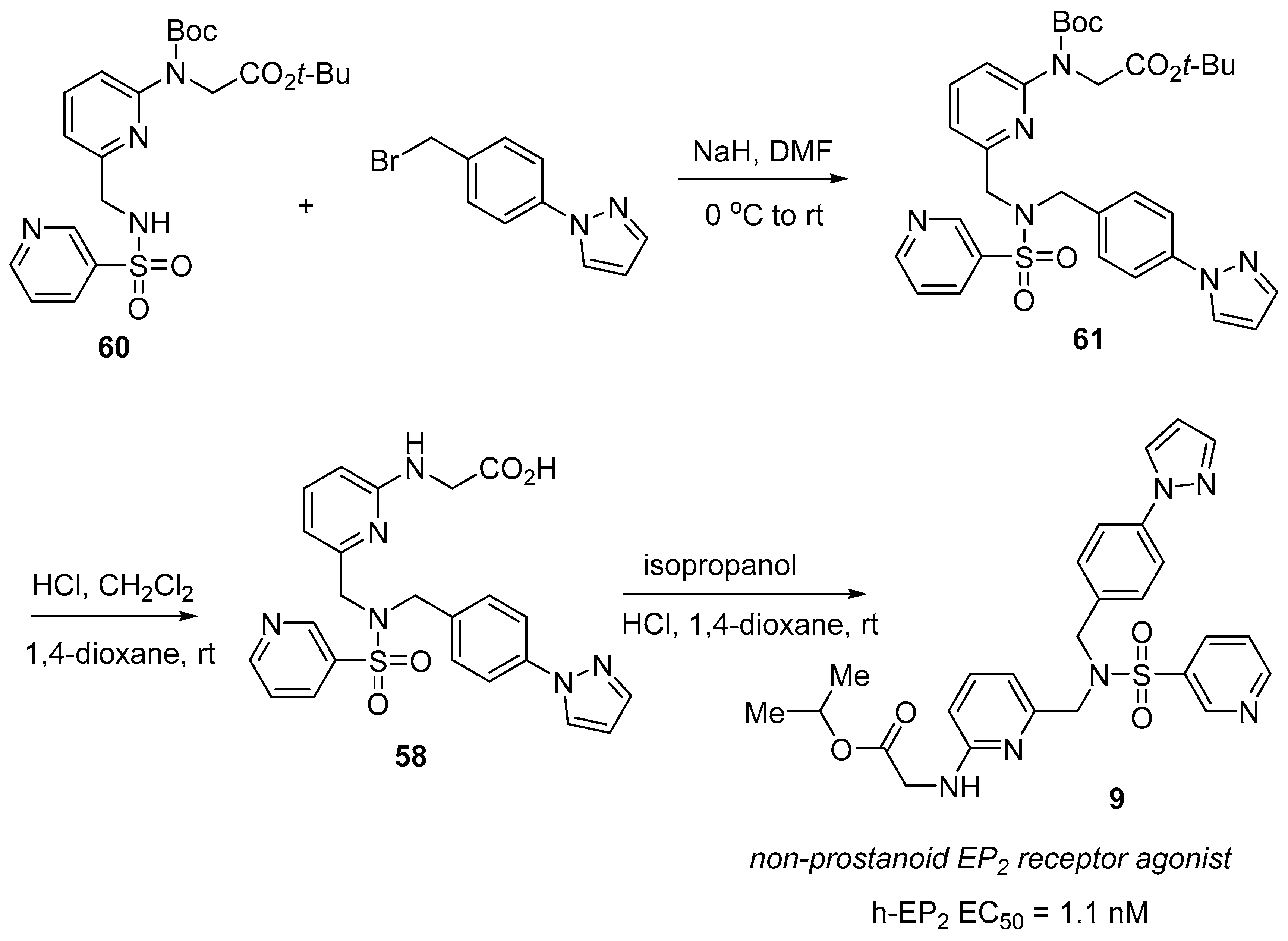
3.5. Omidenepag Isopropyl (OmlontiTM)
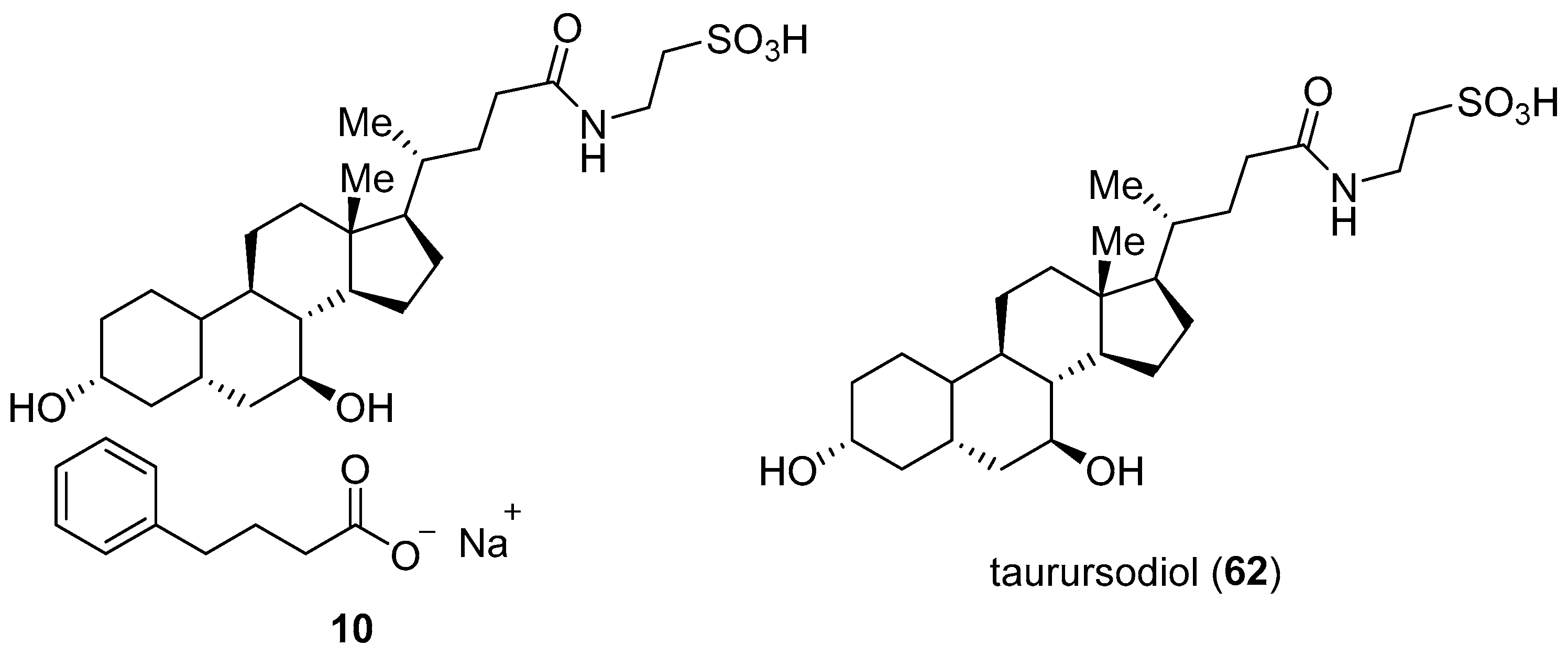
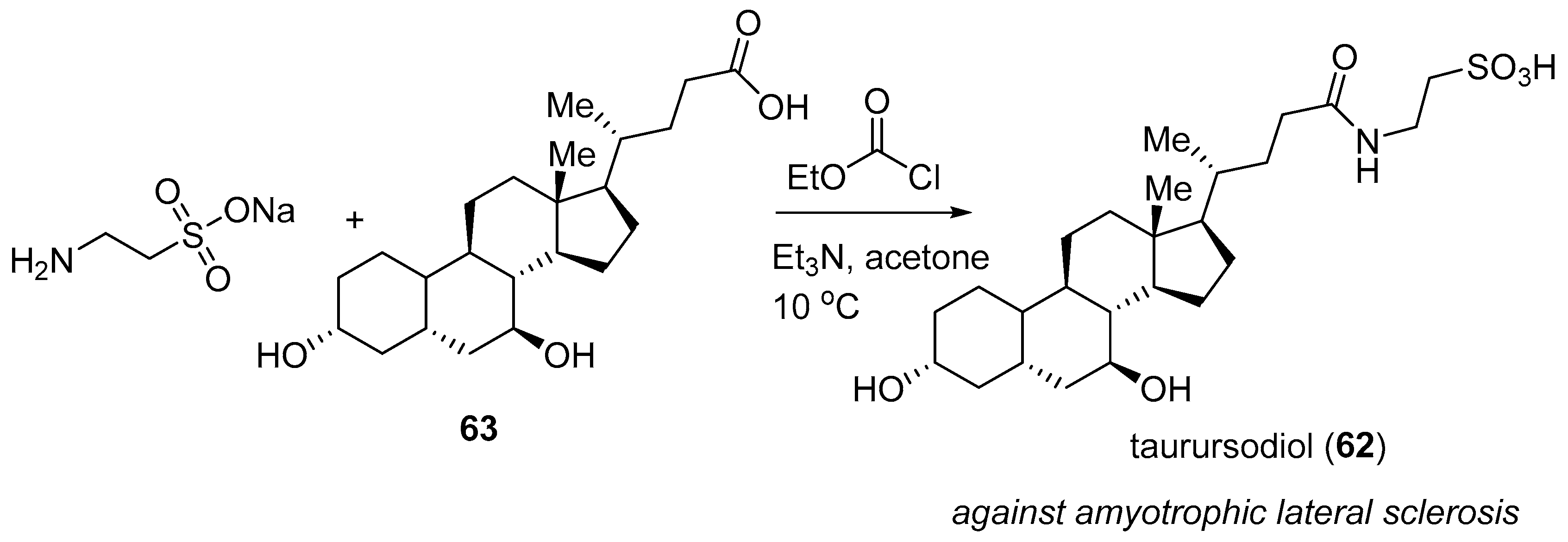
3.6. Phenylbutyrate–Taurursodiol (RelyvrioTM)
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Mei, H.; Han, J.; White, S.; Graham, D.J.; Izawa, K.; Sato, T.; Fustero, S.; Meanwell, N.A.; Soloshonok, V.A. Tailor-Made Amino Acids and Fluorinated Motifs as Prominent Traits in the Modern Pharmaceuticals. Chem. Eur. J. 2020, 26, 11349–11390. [Google Scholar] [CrossRef]
- Wang, Q.; Han, J.; Sorochinsky, A.; Landa, A.; Butler, G.; Soloshonok, V.A. The Latest FDA-Approved Pharmaceuticals Containing Fragments of Tailor-Made Amino Acids and Fluorine. Pharmaceuticals 2022, 15, 999. [Google Scholar] [CrossRef] [PubMed]
- Aboul-Fadl, T.; EI-Shorbagi, A. New prodrug approach for amino acids and amino-acid-like drugs. Eur. J. Med. Chem. 1996, 31, 165–169. [Google Scholar] [CrossRef]
- Blaskovich, M.A.T. Unusual amino acids in medicinal chemistry. J. Med. Chem. 2016, 59, 10807–10836. [Google Scholar] [CrossRef] [PubMed]
- Henninot, A.; Collins, J.C.; Nuss, J.M. The current state of peptide drug discovery: Back to the future? J. Med. Chem. 2018, 61, 1382–1414. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, D.R.W.; Sanderson, J.M. The synthesis of peptides and proteins containing non-natural amino acids. Chem. Soc. Rev. 2004, 33, 422–430. [Google Scholar] [CrossRef]
- Kasten, G.; Grohganz, H.; Rades, T.; Löbmann, K. Development of a screening method for co-amorphous formulations of drugs and amino acids. Eur. J. Pharm. Sci. 2016, 95, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, S. Cyclic peptide drugs approved in the last two decades (2001–2021). RSC Chem. Biol. 2022, 3, 18–31. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Sorochinsky, A.E. Practical Methods for the Synthesis of Symmetrically α,α-Disubstituted-α-Amino Acids. Synthesis 2010, 2010, 2319–2344. [Google Scholar] [CrossRef]
- Han, J.; Sorochinsky, A.E.; Ono, T.; Soloshonok, V.A. Biomimetic Transamination—A Metal-Free Alternative to the Reduc-tive Amination. Application for Generalized Preparation of Fluorine-Containing Amines and Amino Acids. Curr. Org. Synth. 2011, 8, 281–294. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Cai, C.; Hruby, V.J.; Meervelt, L.V. Asymmetric Synthesis of Novel Highly Sterically Constrained (2S,3S)-3-Methyl-3-Trifluoromethyl- and (2S,3S,4R)-3-Trifluoromethyl-4-Methylpyroglutamic Acids. Tetrahedron 1999, 55, 12045–12058. [Google Scholar] [CrossRef]
- Liu, A.; Han, J.; Nakano, A.; Konno, H.; Moriwaki, H.; Abe, H.; Izawa, K.; Soloshonok, V.A. New pharmaceuticals approved by FDA in 2020: Small-molecule drugs derived from amino acids and related compounds. Chirality 2022, 34, 86–103. [Google Scholar] [CrossRef]
- Cjunico, W.; Gomes, C.R.B.; Ferreira, M.L.G.; Ferreira, T.G.; Cardinot, D.; de Souza, M.V.N.; Lourenço, M.C.S. Synthesis and anti-mycobacterial activity of novel amino alcohol derivatives. Eur. J. Med. Chem. 2011, 46, 974–978. [Google Scholar] [CrossRef]
- Han, J.; Konno, H.; Sato, T.; Soloshonok, V.A.; Izawa, K. Tailor-made amino acids in the design of small-molecule blockbuster drugs. Eur. J. Med. Chem. 2021, 220, 113448. [Google Scholar] [CrossRef]
- Clarkson, C.; Musonda, C.C.; Chibale, K.; Campbell, W.E.; Smith, P. Synthesis of totarol amino alcohol derivatives and their antiplasmodial activity and cytotoxicity. Bioorg. Med. Chem. 2003, 11, 4417–4422. [Google Scholar] [CrossRef]
- Liu, J.; Han, J.; Izawa, K.; Sato, T.; White, S.; Meanwell, N.A.; Soloshonok, V.A. Cyclic tailor-made amino acids in the design of modern pharmaceuticals. Eur. J. Med. Chem. 2020, 208, 112736. [Google Scholar] [CrossRef]
- Puris, E.; Gynther, M.; Auriola, S.; Huttunen, K.M. L-Type amino acid transporter 1 as a target for drug delivery. Pharm. Res. 2020, 37, 88. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Han, J.; Klika, K.D.; Izawa, K.; Sato, T.; Meanwell, N.A.; Soloshonok, V.A. Applications of Fluorine-Containing Amino Acids for Drug Design. Eur. J. Med. Chem. 2020, 186, 111826. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D.; Harper, D.B. Fluorine-containing natural products. J. Fluorine Chem. 1999, 100, 127–133. [Google Scholar] [CrossRef]
- O’Hagan, D.; Schaffrath, C.; Cobb, S.L.; Hamilton, J.T.G.; Cormac, C.D. Biosynthesis of an organofluorine molecule. Nature 2002, 416, 279. [Google Scholar] [CrossRef]
- Dong, C.; Huang, F.; Deng, H.; Schaffrath, C.; Spencer, J.B.; O’Hagan, D.; Naismith, J.H. Crystal structure and mechanism of a bacterial fluorinating enzyme. Nature 2004, 427, 561. [Google Scholar] [CrossRef]
- Fried, J.; Sabo, E.F. Synthesis of 17α-hydroxycorticosterone and its 9α-halo derivatives from 11-epi-17α-hydroxycorticosterone. J. Am. Chem. Soc. 1953, 75, 2273–2274. [Google Scholar] [CrossRef]
- Fried, J.; Sabo, E.F. 9α-Fluoro derivatives of cortisone and hydrocortisone. J. Am. Chem. Soc. 1954, 76, 1455–1456. [Google Scholar] [CrossRef]
- Heidelberger, C.; Chaudhuri, N.K.; Danneberg, P.; Mooren, D.; Griesbach, L.; Duschinsky, R.; Schnitzer, R.J.; Pleven, E.; Scheiner, J. Fluorinated pyrimidines, a new class of tumour-inhibitory compounds. Nature 1957, 179, 663–666. [Google Scholar] [CrossRef]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of Fluorine in Medicinal Chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Han, J.L.; Wang, J.; Shibata, N.; Sodeoka, M.; Soloshonok, V.A.; Coelho, J.A.S.; Toste, F.D. Modern Approaches for Asymmetric Construction of Carbon−Fluorine Quaternary Stereogenic Centers: Synthetic Challenges and Pharmaceutical Needs. Chem. Rev. 2018, 118, 3887–3964. [Google Scholar] [CrossRef]
- Han, J.; Kiss, L.; Mei, H.; Remete, A.M.; Ponikvar-Svet, M.; Sedgwick, D.M.; Roman, R.; Fustero, S.; Moriwaki, H.; Soloshonok, V.A. Chemical Aspects of Human and Environmental Overload with Fluorine. Chem. Rev. 2021, 121, 4678–4742. [Google Scholar] [CrossRef]
- Mei, H.; Remete, A.M.; Zou, Y.; Moriwaki, H.; Fustero, S.; Kiss, L.; Soloshonok, V.A.; Han, J. Fluorine-containing drugs ap-proved by the FDA in 2019. Chin. Chem. Lett. 2020, 31, 2401–2413. [Google Scholar] [CrossRef]
- He, J.; Li, Z.; Dhawan, G.; Zhang, W.; Sorochinsky, A.E.; Butler, G.; Soloshonok, V.A.; Han, J. Fluorine-containing drugs approved by the FDA in 2021. Chin. Chem. Lett. 2023, 34, 107578. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, A.; Dhawan, G.; Mei, H.; Zhang, W.; Izawa, K.; Soloshonok, V.A.; Han, J. Fluorine-containing pharmaceuticals approved by the FDA in 2020: Synthesis and biological activity. Chin. Chem. Lett. 2021, 32, 3342–3354. [Google Scholar] [CrossRef]
- Mei, H.; Han, J.; Fustero, S.; Medio-Simon, M.; Sedgwick, D.M.; Santi, C.; Ruzziconi, R.; Soloshonok, V.A. Fluorine-containing drugs approved by the FDA in 2018. Chem. Eur. J. 2019, 25, 11797–11819. [Google Scholar] [CrossRef]
- Bos, J.L. Ras Oncogenes in Human Cancer: A Review Cancer Research. Cancer Res. 1989, 49, 4682–4689. [Google Scholar]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRASG12C Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [PubMed]
- Fell, J.B.; Fischer, J.P.; Baer, B.R.; Ballard, J.; Blake, J.F.; Bouhana, K.; Brandhuber, B.J.; Briere, D.M.; Burgess, L.E.; Burkard, M.R.; et al. Discovery of tetrahydropyridopyrimidines as irreversible covalent inhibitors of KRAS-G12C with in vivo activity. ACS Med. Chem. Lett. 2018, 9, 1230–1234. [Google Scholar] [CrossRef]
- Fell, J.B.; Fischer, J.P.; Baer, B.R.; Blake, J.F.; Bouhana, K.; Briere, D.M.; Brown, K.D.; Burgess, L.E.; Burns, A.C.; Burkard, M.R.; et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 6679–6693. [Google Scholar] [CrossRef] [PubMed]
- Thein, K.Z.; Biter, A.B.; Hong, D.S. Therapeutics targeting mutant KRAS. Ann. Rev. Med. 2021, 72, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Christensen, J.G.; Olson, P.; Briere, T.; Wiel, C.; Bergo, M.O. Targeting Krasg12c-mutant cancer with a mutation-specific inhibitor. J. Int. Med. 2020, 288, 183–191. [Google Scholar] [CrossRef]
- Dunnett-Kane, V.; Nicola, P.; Blackhall, F.; Lindsay, C. Mechanisms of Resistance to KRASG12C Inhibitors. Cancers 2021, 13, 151. [Google Scholar] [CrossRef]
- Jänne, P.A.; Riely, G.J.; Gadgeel, S.M.; Heist, R.S.; Ou, S.I.; Pacheco, J.M.; Johnson, M.L.; Sabari, J.K.; Leventakos, K.; Yau, E.; et al. Adagrasib in Non–Small-Cell Lung Cancer Harboring a KRASG12C Mutation. N. Eng. J. Med. 2022, 387, 120–131. [Google Scholar] [CrossRef]
- Blair, H.A. Sotorasib: First approval. Drugs 2021, 81, 1573–1579. [Google Scholar] [CrossRef]
- Segal-Maurer, S.; DeJesus, E.; Stellbrink, H.-J.; Castagna, A.; Richmond, G.J.; Sinclair, G.I.; Siripassorn, K.; Ruane, P.J.; Berhe, M.; Wang, H.; et al. Capsid inhibition with lenacapavir in multidrug-resistant HIV-1 infection. N. Engl. J. Med. 2022, 386, 1793–1803. [Google Scholar] [CrossRef]
- Thenin-Houssier, S.; Valente, S.T. HIV-1 capsid inhibitors as antiretroviral agents. Curr. HIV Res. 2016, 14, 270–282. [Google Scholar] [CrossRef]
- Carnes, S.K.; Sheehan, J.H.; Aiken, C. Inhibitors of the HIV-1 capsid, a target of opportunity. Curr. Opin. HIV AIDS 2018, 13, 359–365. [Google Scholar] [CrossRef]
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small molecules, big targets: Drug discovery faces the protein–protein interaction challenge. Nat. Rev. Drug Discov. 2016, 15, 533–550. [Google Scholar] [CrossRef]
- Freed, E.O. HIV-1 assembly, release and maturation. Nat. Rev. Microbiol. 2015, 13, 484–496. [Google Scholar] [CrossRef]
- Ganser, B.K.; Li, S.; Klishko, V.Y.; Finch, J.T.; Sundquist, W.I. Assembly and analysis of conical models for the HIV-1 core. Science 1999, 283, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Engelman, A.N. Capsid-dependent host factors in HIV-1 infection. Trends Microbiol. 2017, 25, 741–755. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.T.; Summers, B.J.; Xu, C.; Perilla, J.R.; Malikov, V.; Naghavi, M.H.; Xiong, Y. FEZ1 is recruited to a conserved cofactor site on capsid to promote HIV-1 trafficking. Cell Rep. 2019, 28, 2373–2385. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, J.; Machado, A.K.; Lyonnais, S.; Chamontin, C.; Gärtner, K.; Léger, T.; Henriquet, C.; Garcia, C.; Portilho, D.M.; Pugnière, M.; et al. Transportin-1 binds to the HIV-1 capsid via a nuclear localization signal and triggers uncoating. Nat. Microbiol. 2019, 4, 1840–1850. [Google Scholar] [CrossRef]
- FDA Approves New HIV Drug for Adults with Limited Treatment Options; U.S. Food and Drug Administration (FDA): Rockville, MD, USA, 2022.
- Zhuang, S.; Torbett, B.E. Interactions of HIV-1 capsid with host factors and their implications for developing novel therapeutics. Viruses 2021, 13, 417. [Google Scholar] [CrossRef] [PubMed]
- Link, J.O.; Rhee, M.S.; Tse, W.C.; Zheng, J.; Somoza, J.R.; Rowe, W.; Begley, R.; Chiu, A.; Mulato, A.; Hansen, D.; et al. Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature 2020, 584, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Bester, S.M.; Wei, G.; Zhao, H.; Adu-Ampratwum, D.; Iqbal, N.; Courouble, V.V.; Francis, A.C.; Annamalai, A.S.; Singh, P.K.; Shkriabai, N.; et al. Structural and mechanistic bases for a potent HIV-1 capsid inhibitor. Science 2020, 370, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Gallazzi, F.; Hill, K.J.; Burke, D.H.; Lange, M.J.; Quinn, T.P.; Neogi, U.; Sönnerborg, A. GS-CA compounds: First-in-class HIV-1 capsid inhibitors covering multiple grounds. Front. Microbiol. 2019, 10, 1227. [Google Scholar] [CrossRef] [PubMed]
- Margot, N.; Ram, R.; Rhee, M.; Callebaut, C. Absence of Lenacapavir (GS-6207) Phenotypic Resistance in HIV Gag Cleavage Site Mutants and in Isolates with Resistance to Existing Drug Classes. Antimicrob. Agents Chemother. 2021, 65, e02057-20. [Google Scholar] [CrossRef] [PubMed]
- Graupe, M.; Henry, S.J.; Link, J.O.; Rowe, C.W.; Saito, R.D.; Schroeder, C.S.; Stefanidis, D.; Tse, W.C.; Zhang, J.R. Therapeutic Compounds Useful for the Prophylactic or Therapeutic Treatment of an HIV Virus Infection. WO2018035359A1, 22 February 2018. [Google Scholar]
- Hoy, S.M. Oteseconazole: First approval. Drugs 2022, 82, 1017–1023. [Google Scholar] [CrossRef]
- Warrilow, A.G.S.; Hull, C.M.; Parker, J.E.; Garvey, E.P.; Hoekstra, W.J.; Moore, W.R.; Schotzinger, R.J.; Kelly, D.E.; Kelly, S.L. The clinical candidate VT-1161 is a highly potent inhibitor of Candida albicans CYP51 but fails to bind the human enzyme. Antimicrob. Agents Chemother. 2014, 58, 7121–7127. [Google Scholar] [CrossRef]
- Garvey, E.P.; Hoekstra, W.J.; Moore, W.R.; Schotzinger, R.J.; Long, L.; Ghannoum, M.A. VT-1161 dosed once daily or once weekly exhibits potent efficacy in treatment of dermatophytosis in a guinea pig model. Antimicrob. Agents Chemother. 2015, 59, 1992–1997. [Google Scholar] [CrossRef]
- Martens, M.G.; Maximos, B.; Degenhardt, T.; Person, K.; Curelop, S.; Ghannoum, M.; Flynt, A.; Brand, S.R. Phase 3 study evaluating the safety and efficacy of oteseconazole in the treatment of recurrent vulvovaginal candidiasis and acute vulvovaginal candidiasis infections. Am. J. Obstet. Gynecol. 2022, 227, 880.e1–880.e11. [Google Scholar] [CrossRef]
- Hoekstra, W.J.; Garvey, E.P.; Moore, W.R.; Rafferty, S.W.; Yates, C.M.; Schotzinger, R.J. Design and optimization of highly-selective fungal CYP51 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 3455–3458. [Google Scholar] [CrossRef]
- Wirth, D.D.; Yates, C.M.; Hoekstra, W.J.; Bindl, M.F.; Hartmann, E. Antifungal Compound Process. WO2017049080A1, 23 March 2017. [Google Scholar]
- Garnock-Jones, K.P. Vonoprazan: First Global Approval. Drug 2015, 75, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Shichijo, S.; Hirata, Y.; Niikura, R.; Hayakawa, Y.; Yamada, A.; Mochizuki, S.; Matsuo, K.; Isomura, Y.; Seto, M.; Suzuki, N.; et al. Vonoprazan versus conventional proton pump inhibitor-based triple therapy as first-line treatment against Helicobacter pylori: A multicenter retrospective study in clinical practice. J. Digest. Dis. 2016, 17, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, H.; Sakurai, Y.; Nishimura, A.; Okamoto, H.; Hibberd, M.; Jenkins, R.; Yoneyama, T.; Ashida, K.; Ogama, Y.; Warrington, S. Randomised clinical trial: Safety, tolerability, pharmacokinetics and pharmacodynamics of repeated doses of TAK-438 (vonoprazan), a novel potassium-competitive acid blocker, in healthy male subjects. Aliment. Pharmacol. Ther. 2015, 41, 636–648. [Google Scholar] [CrossRef]
- Huh, K.Y.; Chung, H.; Kim, Y.K.; Lee, S.H.; Bhatia, S.; Takanami, Y.; Nakaya, R.; Yu, K.S. Evaluation of safety and pharmacokinetics of bismuth–containing quadruple therapy with either vonoprazan or lansoprazole for Helicobacter pylori eradication. Br. J. Clin. Pharmacol. 2022, 88, 138–144. [Google Scholar] [CrossRef]
- Kagami, T.; Sahara, S.; Ichikawa, H.; Uotani, T.; Yamade, M.; Sugimoto, M.; Hamaya, Y.; Iwizumi, M.; Osawa, S.; Sugimoto, K.; et al. Potent acid inhibition by vonoprazan in comparison with esomeprazole, with reference to CYP 2C19 genotype. Aliment. Pharmacol. Ther. 2016, 43, 1048–1059. [Google Scholar] [CrossRef]
- Murakami, K.; Sakurai, Y.; Shiino, M.; Funao, N.; Nishimura, A.; Asaka, M. Vonoprazan, a novel potassium-competitive acid blocker, as a component of first-line and second-line triple therapy for Helicobacter pylori eradication: A phase III, randomised, double-blind study. Gut 2016, 65, 1439–1446. [Google Scholar] [CrossRef]
- Arikawa, Y.; Nishida, H.; Kurasawa, O.; Hasuoka, A.; Hirase, K.; Inatomi, N.; Hori, Y.; Matsukawa, J.; Imanishi, A.; Kondo, M.; et al. Discovery of a Novel Pyrrole Derivative 1-[5-(2-Fluorophenyl)-1-(pyridin-3-ylsulfonyl)-1H-pyrrol-3-yl]-N-methylmethanamine Fumarate (TAK-438) as a Potassium-Competitive Acid Blocker (P-CAB). J. Med. Chem. 2012, 55, 4446–4456. [Google Scholar] [CrossRef] [PubMed]
- Urbanová, K.; Seifert, D.; Vinšová, H.; Vlk, M.; Lebeda, O. Simple new method for labelling of PSMA-11 with 68Ga in NaHCO3. Appl. Radiat. Isot. 2021, 172, 109692. [Google Scholar] [CrossRef]
- Sheikhbahaei, S.; Werner, R.A.; Solnes, L.B.; Pienta, K.J.; Pomper, M.G.; Gorin, M.A.; Rowe, S.P. Prostate-Specific Membrane Antigen (PSMA)-Targeted pet imaging of prostate cancer: An update on important pitfalls. Semin. Nucl. Med. 2019, 49, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Debnath, S.; Zhou, N.; McLaughlin, M.; Rice, S.; Pillai, A.K.; Hao, G.; Sun, X. PSMA-targeting imaging and theranostic agents Current status and future perspective. Int. J. Mol. Sci. 2022, 23, 1158. [Google Scholar] [CrossRef]
- Horoszewicz, J.S.; Kawinski, E.; Murphy, G.P. Monoclonal antibodies to a new antigenic marker in epithelial prostatic cells and serum of prostatic cancer patients. Anticancer Res. 1987, 7, 927–935. [Google Scholar] [PubMed]
- Sartor, O.; de Bono, J.; Chi, K.N.; Fizazi, K.; Herrmann, K.; Rahbar, K.; Tagawa, S.T.; Nordquist, L.T.; Vaishampayan, N.; El-Haddad, G.; et al. Lutetium-177-PSMA-617 for Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2021, 385, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Benešová, M.; Schäfer, M.; Bauder-Wüst, U.; Afshar-Oromieh, A.; Kratochwil, C.; Mier, W.; Haberkorn, U.; Kopka, K.; Eder, M. Preclinical Evaluation of a Tailor-Made DOTA-Conjugated PSMA Inhibitor with Optimized Linker Moiety for Imaging and Endoradiotherapy of Prostate Cancer. J. Nucl. Med. 2015, 56, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Eder, M.; Schäfer, M.; Bauder-Wüst, U.; Hull, W.-E.; Wängler, C.; Mier, W.; Haberkorn, U.; Eisenhut, M. 68 Ga-Complex Lipophilicity and the Targeting Property of a Urea-Based PSMA Inhibitor for PET Imaging. Bioconjugate Chem. 2012, 23, 688–697. [Google Scholar] [CrossRef]
- Green, E.M.; Wakimoto, H.; Anderson, R.L.; Evanchik, M.J.; Gorham, J.M.; Harrison, B.C.; Henze, M.; Kawas, R.; Oslob, J.D.; Rodriguez, H.M.; et al. A small molecule inhibitor of sarcomere contractility suppresses hypertrophic cardiomyopathy in mice. Science 2016, 351, 617–621. [Google Scholar] [CrossRef]
- Anderson, R.L.; Trivedi, D.V.; Sarkar, S.S.; Henze, M.; Ma, W.; Gong, H.; Rogers, C.S.; Gorham, J.M.; Wong, F.L.; Morck, M.M.; et al. Deciphering the super relaxed state of human β-cardiac myosin and the mode of action of mavacamten from myosin molecules to muscle fibers. Proc. Natl. Acad. Sci. USA 2018, 115, E8143–E8152. [Google Scholar] [CrossRef]
- Toepfer, C.N.; Wakimoto, H.; Garfinkel, A.C.; Mcdonough, B.; Liao, D.; Jiang, J.; Tai, A.C.; Gorham, J.M.; Lunde, I.G.; Lun, M.; et al. Hypertrophic cardiomyopathy mutations in MYBPC3 dysregulate myosin. Sci. Transl. Med. 2019, 11, eaat1199. [Google Scholar] [CrossRef]
- Mamidi, R.; Li, J.; Doh, C.Y.; Verma, S.; Stelzer, J.E. Impact of the myosin modulator mavacamten on force generation and cross-bridge behavior in a murine model of hypercontractility. J. Am. Heart Assoc. 2018, 7, e009627. [Google Scholar] [CrossRef]
- Awinda, P.O.; Bishaw, Y.; Watanabe, M.; Guglin, M.A.; Campbell, K.S.; Tanner, B.C.W. Effects of mavacamten on Ca2+ sensitivity of contraction as sarcomere length varied in human myocardium. Br. J. Pharmacol. 2020, 177, 5609–5621. [Google Scholar] [CrossRef]
- Keam, S.J. Mavacamten: First Approval. Drugs 2022, 82, 1127–1135. [Google Scholar] [CrossRef]
- Oslob, J.; Anderson, R.; Aubele, D.; Evanchik, M.; Fox, J.C.; Kane, B.; Lu, P.; McDowell, R.; Rodriguez, H.; Song, Y.; et al. Pyrimidinedione Compounds. US 9585883 B2, 7 March 2017. [Google Scholar]
- Ziemichód, W.; Grabowska, K.; Kurowska, A.; Biała, G. A Comprehensive Review of Daridorexant, a Dual-Orexin Receptor Antagonist as New Approach for the Treatment of Insomnia. Molecules 2022, 27, 6041. [Google Scholar] [CrossRef] [PubMed]
- Boss, C.; Gatfield, J.; Brotschi, C.; Heidmann, B.; Sifferlen, T.; von Raumer, M.; Schmidt, G.; Williams, J.T.; Treiber, A.; Roch, C. The Quest for the Best Dual Orexin Receptor Antagonist (Daridorexant) for the Treatment of Insomnia Disorders. ChemMedChem 2020, 15, 2286–2305. [Google Scholar] [CrossRef] [PubMed]
- Mignot, E.; Mayleben, D.; Fietze, I.; Leger, D.; Zammit, G.; Bassetti, C.L.A.; Pain, S.; Kinter, D.S.; Roth, T. Safety and efficacy of daridorexant in patients with insomnia disorder: Results from two multicentre, randomised, double-blind, placebo-controlled, phase 3 trials. Lancet Neurol. 2022, 21, 125–139. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Daridorexant: First Approval. Drugs 2022, 82, 601–607. [Google Scholar] [CrossRef]
- Boss, C.; Brotschi, C.; Gude, M.; Heidmann, B.; Sifferlen, T.; von Raumer, M.; Williams, J.T. Crystalline Form of (S)-(2-(6-Chloro-7-methyl-1H-benzo[d]imidazol-2-yl)-2-methylpyrrolidin-1-yl)(5-methoxy-2-(2H-1,2,3-triazol-2-yl)phenyl)methanone and Its Use as Orexin Receptor Antagonists. WO2015083070, 11 June 2015. [Google Scholar]
- Robic, C.; Port, M.; Rousseaux, O.; Louguet, S.; Fretellier, N.; Catoen, S. Physicochemical and pharmacokinetic profiles of gadopiclenol: A new macrocyclic gadolinium chelate with high T1 relaxivity. Investig. Radiol. 2019, 54, 475–484. [Google Scholar] [CrossRef]
- Port, M. Compounds Comprising Short Aminoalcohol Chains and Metal Complexes for Medical Imaging. EP1931673 B1, 29 August 2012. [Google Scholar]
- Napolitano, R.; Lattuada, L.; Baranyai, Z.; Guidolin, N.; Marazzi, G. Gadolinium Bearing Pcta-Based Contrast Agents. WO2020030618A1, 13 February 2020. [Google Scholar]
- Gauvreau, G.M.; Watson, R.M.; O’Byrne, P.M. Protective Effects of Inhaled PGE2 on Allergen-induced Airway Responses and Airway Inflammation. Am. J. Respir. Crit. Care Med. 1999, 159, 31–36. [Google Scholar] [CrossRef]
- Suzawa, T.; Miyaura, C.; Inada, M.; Maruyama, T.; Sugimoto, Y.; Ushikubi, F.; Ichikawa, A.; Narumiya, S.; Suda, T. The role of prostaglandin E receptor subtypes (EP1, EP2, EP3, and EP4) in bone resorption: An analysis using specific agonists for the respective Eps. Endocrinology 2000, 141, 1554–1559. [Google Scholar] [CrossRef]
- Flach, A.J.; Eliason, J.A. Topical Prostaglandin E2 Effects on Normal Human Intraocular Pressure. J. Ocular Pharmacol. Ther. 1988, 4, 13–18. [Google Scholar] [CrossRef]
- Sugimoto, Y.; Narumiya, S. Prostaglandin E receptors. J. Biol. Chem. 2007, 282, 11613–11617. [Google Scholar] [CrossRef]
- Ganesh, T. Prostanoid receptor EP2 as a therapeutic target: Miniperspective. J. Med. Chem. 2014, 57, 4454–4465. [Google Scholar] [CrossRef]
- Cameron, K.O.; Lefker, B.A.; Rosati, R.L. Prostaglandin Agonists and Their Use to Treat Bone Disorders. WO1999019300, 22 April 1999. [Google Scholar]
- Paralkar, V.M.; Borovecki, F.; Ke, H.Z.; Cameron, K.O.; Lefker, B.; Grasser, W.A.; Owen, T.A.; Li, M.; DaSilva-Jardine, P.; Zhou, M.; et al. An EP2 receptor-selective prostaglandin E2 agonist induces bone healing. Proc. Natl. Acad. Sci. USA 2003, 100, 6736–6740. [Google Scholar] [CrossRef] [PubMed]
- Constan, A.A.; Keshary, P.R.; Maclean, D.B.; Paralkar, V.M.; Roman, D.C.; Thompson, D.D.; Wright, T.M. Use of EP2 Selective Receptor Agonists in Medicinal Treatment. WO2004078169, 16 September 2004. [Google Scholar]
- Williams, D.H.; Stephens, E.; O’Brien, D.P.; Zhou, M. Understanding noncovalent interactions: Ligand binding energy and catalytic efficiency from ligand-induced reductions in motion within receptors and enzymes. Angew. Chem. Int. Ed. 2004, 43, 6596–6616. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, F.; Vaidehi, N.; Goddard, W.A.; Sheinerman, F.; Reiling, S.; Morize, I.; Mu, L.; Harris, K.; Ardati, A.; et al. Prediction of the 3D structure and dynamics of human DP G-protein coupled receptor bound to an agonist and an antagonist. J. Am. Chem. Soc. 2007, 129, 10720–10731. [Google Scholar] [CrossRef]
- Lach, J.L.; Huang, H.S.; Schoenwald, R.D. Corneal penetration behavior of β-blocking agents II: Assessment of barrier contributions. J. Pharm. Sci. 1983, 72, 1272–1279. [Google Scholar] [CrossRef]
- Rojanasakul, Y.; Robinson, J.R. Transport mechanisms of the cornea: Characterization of barrier permselectivity. Int. J. Pharm. 1989, 55, 237–246. [Google Scholar] [CrossRef]
- Järvinen, T.; Järvinen, K. Prodrugs for improved ocular drug delivery. Adv. Drug Deliver. Rev. 1996, 19, 203–224. [Google Scholar] [CrossRef]
- Prasanna, G.; Bosworth, C.F.; Lafontaine, J.A. EP2 Agonists. WO2008015517, 7 February 2008. [Google Scholar]
- Cheng-Bennett, A.; Chan, M.F.; Chen, G.; Gac, T.; Garst, M.E.; Gluchowski, C. Studies on a novel series of acyl ester prodrugs of prostaglandin F2 alpha. Br. J. Ophthalmol. 1994, 78, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Kirihara, T.; Taniguchi, T.; Yamamura, K.; Iwamura, R.; Yoneda, K.; Odani-Kawabata, N.; Shimazaki, A.; Matsugi, T.; Shams, N.; Zhang, J.Z. Pharmacologic characterization of omidenepag isopropyl, a novel selective EP2 receptor agonist, as an ocular hypotensive agent. Investig. Ophthalmol. Vis. Sci. 2018, 59, 145–153. [Google Scholar] [CrossRef]
- Iwamura, R.; Tanaka, M.; Okanari, E.; Kirihara, T.; Odani-Kawabata, N.; Shams, N.; Yoneda, K. Identification of a selective, non-prostanoid EP2 receptor agonist for the treatment of glaucoma: Omidenepag and its prodrug omidenepag isopropyl. J. Med. Chem. 2018, 61, 6869–6891. [Google Scholar] [CrossRef]
- Traxinger, K.; Kelly, C.; Johnson, B.A.; Lyles, R.H.; Glass, J.D. Prognosis and epidemiology of amyotrophic lateral sclerosis: Analysis of a clinic population, 1997–2011. Neurol. Clin. Pract. 2013, 3, 313–320. [Google Scholar] [CrossRef]
- Jaronen, M.; Goldsteins, G.; Koistinaho, J. ER stress and unfolded protein response in amyotrophic lateral sclerosis—A controversial role of protein disulphide isomerase. Front. Cell Neurosci. 2014, 8, 402. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.R.; Walters, R.; Waldron, F.; Pal, S.; Selvaraj, B.T.; MacLeod, M.R.; Hardingham, G.E.; Chandran, S.; Gregory, J.M. Targeting mitochondrial dysfunction in amyotrophic lateral sclerosis: A systematic review and meta-analysis. Brain Commun. 2019, 1, fcz009. [Google Scholar] [CrossRef]
- Iannitti, T.; Palmieri, B. Clinical and experimental applications of sodium phenylbutyrate. Drugs R&D 2011, 11, 227–249. [Google Scholar]
- Walker, V. Ammonia toxicity and its prevention in inherited defects of the urea cycle. Diabetes Obes. Metab. 2009, 11, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Kaur, B.; Bhat, A.; Chakraborty, R.; Adlakha, K.; Sengupta, S.; Roy, S.S.; Chakraborty, K. Proteomic profile of 4-PBA treated human neuronal cells during ER stress. Mol. Omics 2018, 14, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Suaud, L.; Miller, K.; Panichelli, A.E.; Randell, R.L.; Marando, C.M.; Rubenstein, R.C. 4-Phenylbutyrate stimulates Hsp70 expression through the Elp2 component of elongator and STAT-3 in cystic fibrosis epithelial cells. J. Biol. Chem. 2011, 286, 45083–45092. [Google Scholar] [CrossRef]
- Paganoni, S.; Macklin, E.A.; Hendrix, S.; Berry, J.D.; Elliott, M.A.; Maiser, S.; Karam, C.; Caress, J.B.; Owegi, M.A.; Quick, A.; et al. Trial of sodium phenylbutyrate–taurursodiol for amyotrophic lateral sclerosis. N. Engl. J. Med. 2020, 383, 919–930. [Google Scholar] [CrossRef]
- Rodrigues, C.M.P.; Solá, S.; Sharpe, J.C.; Moura, J.J.G.; Steer, C.J. Tauroursodeoxycholic acid prevents Bax-induced membrane perturbation and cytochrome C release in isolated mitochondria. Biochemistry 2003, 42, 3070–3080. [Google Scholar] [CrossRef]
- Brown, A. FDA new drug approvals in Q3 2022. Nat. Rev. Drug Discov. 2022, 21, 788. [Google Scholar] [CrossRef]
- Kang, Y.; Qiu, C.; Gu, H. Preparation Method for Sodium Phenylbutyrate. CN105924345A, 7 September 2016. [Google Scholar]
- Parenti, M. Process for the Preparation of Tauroursodesoxycholic Acid. EP1985622A1, 29 October 2008. [Google Scholar]
- Wang, Y.; Song, X.; Wang, J.; Moriwaki, H.; Soloshonok, V.A.; Liu, H. Recent approaches for asymmetric synthesis of -amino acids via homologation of Ni(II) complexes. Amino Acids 2017, 49, 1487–1520. [Google Scholar] [CrossRef] [PubMed]
- Aceña, J.L.; Sorochinsky, A.E.; Soloshonok, V. Asymmetric synthesis of α-amino acids via homologation of Ni(II) complexes of glycine Schiff bases. Part 3: Michael addition reactions and miscellaneous transformations. Amino Acids 2014, 46, 2047–2073. [Google Scholar] [CrossRef] [PubMed]
- Takeda, R.; Kawamura, A.; Kawashima, A.; Sato, T.; Moriwaki, H.; Izawa, K.; Akaji, K.; Wang, S.; Liu, H.; Aceña, J.L.; et al. Chemical Dynamic Kinetic Resolution and (S)/(R)-Interconversion of Unprotected α-Amino Acids. Angew. Chem. Int. Ed. 2014, 53, 12214–12217. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Wang, J.; Chen, X.; Aceña, J.L.; Soloshonok, V.A.; Liu, H. Chemical Kinetic Resolution of Unprotected b-Substituted-b-Amino Acids Using Recyclable Chiral Ligands. Angew. Chem. Int. Ed. 2014, 53, 7883–7886. [Google Scholar] [CrossRef] [PubMed]
- Tressaud, A.; Haufe, G. (Eds.) Fluorine and Health: Molecular Imaging, Biomedical Materials and Pharmaceuticals; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Kirsch, P. Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, 2nd ed.; Completely Revised and Enlarged; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013. [Google Scholar]
- Ragni, R.; Punzi, A.; Babudri, F.; Farinola, G.M. Organic and organometallic fluorinated materials for electronics and optoelectronics: A survey on recent research. Eur. J. Org. Chem. 2018, 2018, 3500–3519. [Google Scholar] [CrossRef]
- Begue, J.P.; Bonnet-Delpon, D. Bioorganic and Medicinal Chemistry of Fluorine; John Wiley & Sons: Hoboken, NJ, USA, 2008. [Google Scholar]
- Yamada, T.; Okada, T.; Sakaguchi, K.; Ohfune, Y.; Ueki, H.; Soloshonok, V.A. Efficient Asymmetric Synthesis of Novel 4-Substituted and Configurationally Stable Analogs of Thalidomide. Org. Lett. 2006, 8, 5625–5628. [Google Scholar] [CrossRef]
- Röschenthaler, G.V.; Kukhar, V.P.; Kulik, I.B.; Belik, M.Y.; Sorochinsky, A.E.; Rusanov, E.B.; Soloshonok, V.A. Asymmetric synthesis of phosphonotrifluoroalanine and its derivatives using N-tert-butanesulfinyl imine derived from fluoral. Tetrahedron Lett. 2012, 53, 539–542. [Google Scholar] [CrossRef]
- Yerien, D.E.; Barata-Vallejo, S.; Postigo, A. Difluoromethylation reactions of organic compounds. Chem. Eur. J. 2017, 23, 14676–14701. [Google Scholar] [CrossRef]
















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, N.; Mei, H.; Dhawan, G.; Zhang, W.; Han, J.; Soloshonok, V.A. New Approved Drugs Appearing in the Pharmaceutical Market in 2022 Featuring Fragments of Tailor-Made Amino Acids and Fluorine. Molecules 2023, 28, 3651. https://doi.org/10.3390/molecules28093651
Wang N, Mei H, Dhawan G, Zhang W, Han J, Soloshonok VA. New Approved Drugs Appearing in the Pharmaceutical Market in 2022 Featuring Fragments of Tailor-Made Amino Acids and Fluorine. Molecules. 2023; 28(9):3651. https://doi.org/10.3390/molecules28093651
Chicago/Turabian StyleWang, Nana, Haibo Mei, Gagan Dhawan, Wei Zhang, Jianlin Han, and Vadim A. Soloshonok. 2023. "New Approved Drugs Appearing in the Pharmaceutical Market in 2022 Featuring Fragments of Tailor-Made Amino Acids and Fluorine" Molecules 28, no. 9: 3651. https://doi.org/10.3390/molecules28093651
APA StyleWang, N., Mei, H., Dhawan, G., Zhang, W., Han, J., & Soloshonok, V. A. (2023). New Approved Drugs Appearing in the Pharmaceutical Market in 2022 Featuring Fragments of Tailor-Made Amino Acids and Fluorine. Molecules, 28(9), 3651. https://doi.org/10.3390/molecules28093651