
Benzofuran Derivatives from Cortex Mori Radicis and Their Cholinesterase-Inhibitory Activity

 ,
,
Abstract
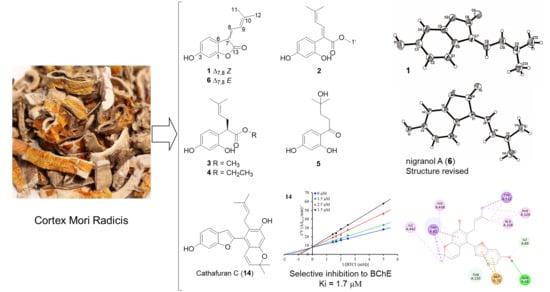
:
1. Introduction
2. Results and Discussion
2.1. Structure Determination of Compounds 1–16
2.2. ChE-Inhibitory Activities
2.3. Molecular Docking and Dynamics Simulation for Cathafuran C with BChE
3. Materials and Methods
3.1. General Experimental Procedure
3.2. Plant Material
3.3. Extraction and Isolation
3.4. ChE Inhibitory Activity Assay
3.5. Kinetic Study of BChE Inhibition
3.6. Molecular Docking and Molecular Dynamics Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Disease International. World Alzheimer Report 2019: Attitudes to Dementia [R]; Alzheimer’s Disease International: London, UK, 2019; pp. 8–9. [Google Scholar]
- Goedert, M.; Spillantini, M.G. A century of Alzheimer’s disease. Science 2006, 314, 777–781. [Google Scholar] [CrossRef]
- Grundke-Iqbal, I.; Iqbal, K.; Tung, Y.C.; Quinlan, M.; Wisniewski, H.M.; and Binder, L.I. Abnormal phosphorylation of the microtubule associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [PubMed]
- Linker, R.A.; Lee, D.H.; Ryan, S.; van Dam, A.M.; Conrad, R.; Bista, P.; Zeng, W.; Hronowsky, X.; Buko, A.; Chollate, S.; et al. Fumaric acid esters exert neuroprotective effects in neuroinflammation via activation of the Nrf2 antioxidant pathway. Brain 2011, 134, 678–692. [Google Scholar] [CrossRef] [PubMed]
- Aliev, G.; Priyadarshini, M.; Reddy, V.P.; Grieg, N.H.; Kaminsky, Y.; Cacabelos, R.; Ashraf, G.M.; Jabir, N.R.; Kamal, M.A.; Nikolenko, V.N.; et al. Oxidative stress mediated mitochondrial and vascular lesions as markers in the pathogenesis of Alzheimer disease. Curr. Med. Chem. 2014, 21, 2208–2217. [Google Scholar] [CrossRef]
- Craig, L.A.; Hong, N.S.; McDonald, R.J. Revisiting the cholinergic hypothesis in the development of Alzheimer’s disease. Neurosci. Biobehav. Rev. 2011, 35, 1397–1409. [Google Scholar] [CrossRef]
- Li, Q.; He, S.; Chen, Y.; Feng, F.; Qu, W.; Sun, H. Donepezil-based multi-functional cholinesterase inhibitors for treatment of Alzheimer’s disease. Eur. J. Med. Chem. 2018, 158, 463–477. [Google Scholar] [CrossRef]
- Li, Q.; Chen, Y.; Xing, S.; Liao, Q.; Xiong, B.; Wang, Y.; Lu, W.; He, S.; Feng, F.; Liu, W.; et al. Highly potent and selective butyrylcholinesterase inhibitors for cognitive improvement and neuroprotection. J. Med. Chem. 2021, 64, 6856–6876. [Google Scholar] [CrossRef]
- Rossi, M.; Freschi, M.; de Camargo Nascente, L.; Salerno, A.; de Melo Viana Teixeira, S.; Nachon, F.; Chantegreil, F.; Soukup, O.; Prchal, L.; Malaguti, M.; et al. Sustainable drug discovery of multi-target-directed ligands for Alzheimer’s Disease. J. Med. Chem. 2021, 64, 4972–4990. [Google Scholar] [CrossRef]
- Xing, S.; Sun, H. The role of butyrylcholinesterase in Alzheimer’s disease and design strategies of its inhibitors. Prog. Pharm. Sci. 2023, 47, 179–193. [Google Scholar]
- Orhan, I.E.; Senol, F.S.; Shekfeh, S.; Skalicka-Wozniak, K.; Banoglu, E. Pteryxin−A promising butyrylcholinesterase-inhibiting coumarin derivative from Mutellina purpurea. Food Chem. Toxicol. 2017, 109, 970–974. [Google Scholar] [CrossRef]
- Jabir, N.R.; Khan, F.R.; and Tabrez, S. Cholinesterase targeting by polyphenols: A therapeutic approach for the treatment of Alzheimer’s disease. CNS Neurosci. Ther. 2018, 24, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Tu, Y.B.; Li, Z.; Li, Y.F. Highly selective carbamate-based butyrylcholinesterase inhibitors derived from a naturally occurring pyranoisoflavone. Bioorganic Chem. 2019, 88, 102949. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Drew, J.; Berney, W.; Lei, W. Neuroprotective natural products for Alzheimer’s disease. Cells 2021, 10, 1309. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Deng, S.; Li, G.; Zhang, Y.; Wang, L.; Wu, C.; Deng, Y. Butenolide derivatives from Aspergillus terreus selectively inhibit butyrylcholinesterase. Front. Chem. 2022, 10, 1063284. [Google Scholar] [CrossRef]
- The State Pharmacopoeia Commission of P., R. China. Pharmacopoeia of People’s Republic of China; The Medicine Science and Technology Press of China: Beijing, China, 2020. [Google Scholar]
- Chan, E.W.C.; Lye, P.Y.; Wong, S.K. Phytochemistry, pharmacology, and clinical trials of Morus alba. Chin. J. Nat. Med. 2016, 14, 17–30. [Google Scholar] [PubMed]
- Wei, H.; Zhu, J.J.; Liu, X.Q.; Feng, W.H.; Wang, Z.M.; Yan, L.H. Review of bioactive compounds from root barks of Morus plants (Sang-Bai-Pi) and their pharmacological effects. Cogent Chem. 2016, 2, 1212320. [Google Scholar] [CrossRef]
- Paudel, P.; Yu, T.; Seong, S.H.; Kuk, E.B.; Jung, H.A.; Choi, J.S. Protein tyrosine phosphatase 1B inhibition and glucose uptake potentials of mulberrofuran G, albanol B, and kuwanon G from root bark of Morus alba L. in insulin-resistant HepG2 cells: An in vitro and in silico study. Int. J. Mol. Sci. 2018, 19, 1542. [Google Scholar] [CrossRef]
- Seong, S.H.; Ha, M.T.; Min, B.S.; Jung, H.A.; Choi, J.S. Moracin derivatives from Morus Radix as dual BACE1 and cholinesterase inhibitors with antioxidant and anti-glycation capacities. Life Sci. 2018, 210, 20–28. [Google Scholar] [CrossRef]
- Paudel, P.; Seong, S.H.; Zhou, Y.; Ha, M.T.; Min, B.S.; Jung, H.A.; Choi, J.S. Arylbenzofurans from the Root Bark of Morus alba as Triple Inhibitors of Cholinesterase, β-Site Amyloid Precursor Protein Cleaving Enzyme 1, and Glycogen Synthase Kinase-3β: Relevance to Alzheimer’s Disease. ACS Omega 2019, 4, 6283–6294. [Google Scholar] [CrossRef]
- Kuk, E.B.; Jo, A.R.; Oh, S.I.; Sohn, H.S.; Seong, S.H.; Roy, A.; Choi, J.S.; Jung, H.A. Anti-Alzheimer’s disease activity of compounds from the root bark of Morus alba L. Arch. Pharmacal Res. 2017, 40, 338–349. [Google Scholar] [CrossRef]
- Xia, C.L.; Tang, G.H.; Guo, Y.Q.; Xu, Y.K.; Huang, Z.S.; Yin, S. Mulberry Diels-Alder-type adducts from Morus alba as multi-targeted agents for Alzheimer’s disease. Phytochemistry 2019, 157, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Yu, M.; Niu, L.; Huang, C.; Wang, Y.; Wu, C.; Yang, P.; Hu, X. Phenolic compounds isolated from Morus nigra and their α-glucosidase inhibitory activities. Nat. Prod. Res. 2020, 34, 605–612. [Google Scholar] [CrossRef] [PubMed]
- Takasugi, M.; Nagao, S.; Masamune, T.; Shirata, A.; Takahashi, K. Structure of moracin A and B, new phytoalexins from diseased mulberry. Tetrahedron Lett. 1978, 19, 797–798. [Google Scholar] [CrossRef]
- Takasugi, M.; Nagao, S.; Ueno, S.; Masamune, T.; Shirata, A.; Takahashi, K. Moracin C and D, new phytoalexins from diseased mulberry. Chem. Lett. 1978, 1978, 1239–1240. [Google Scholar] [CrossRef]
- Zhou, C.X.; Tanaka, J.; Cheng, C.H.; Higa, T.; Tan, R.X. Steroidal alkaloids and stilbenoids from Veratrum taliense. Planta Medica 1999, 65, 480–482. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.H.; Ryu, Y.B.; Curtis-Long, M.J.; Ryu, H.W.; Baek, Y.S.; Kang, J.E.; Lee, W.S.; Park, K.H. Tyrosinase inhibitory polyphenols from roots of Morus lhou. J. Agric. Food Chem. 2009, 57, 1195–1203. [Google Scholar] [CrossRef]
- Kaur, N.; Xia, Y.; Jin, Y.; Dat, N.T.; Gajulapati, K.; Choi, Y.; Hong, Y.S.; Lee, J.J.; Lee, K. The first total synthesis of moracin O and moracin P, and establishment of the absolute configuration of moracin O. Chem. Commun. 2009, 14, 1879–1881. [Google Scholar] [CrossRef]
- Ni, G.; Zhang, Q.J.; Zheng, Z.F.; Chen, R.Y.; Yu, D.Q. 2-Arylbenzofuran Derivatives from Morus cathayana. J. Nat. Prod. 2009, 72, 966–968. [Google Scholar] [CrossRef]
- Nomura, T.; Fukai, T.; Pei, Y.H.; Xu, C.Q.; Chen, Y.J. Components of the root bark of Morus cathayana. 1. Structures of five new isoprenylated flavonoids, sanggenols A-E and a diprenyl-2-arylbenzofuran, mulberrofuran V. Heterocycles 1996, 43, 425–436. [Google Scholar] [CrossRef]
- Hano, Y.; Kohno, H.; Itoh, M.; Nomura, T. Structures of three new 2-arylbenzofuran derivatives from the chinese crude drug ‘sang-bai-pi’ (morus root bark). Chem. Pharm. Bull. 1985, 33, 5294–5300. [Google Scholar] [CrossRef]
- Sakuma, M.; Sakakura, A.; Ishihara, K. Kinetic resolution of racemic carboxylic acids through asymmetric protolactonization promoted by chiral phosphonous acid diester. Org. Lett. 2013, 15, 2838–2841. [Google Scholar] [CrossRef]
- Wu, T.; Zhu, J.X.; Wei, Q.; Li, P.; Wang, L.B.; Huang, J.; Wang, J.H.; Tang, L.K.; Wu, L.J.; Li, C.; et al. Preparative separation of four isomers of synthetic anisodamine by HPLC and diastereomer crystallization. Chirality 2019, 31, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A. Enzymes: A Practical Introduction to Structure, Mechanism, and Data Analysis; Viley-VCH: New York, NY, USA, 2000; pp. 273–277. [Google Scholar]
- Buker, S.M.; Boriack-Sjodin, P.A.; Copeland, R.A. Enzyme-inhibitor interactions and a simple, rapid method for determining inhibition modality. SLAS Discov. 2019, 24, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Nicolet, Y.; Lockridge, O.; Masson, P.; Fontecilla-Camps, J.C.; Nachon, F. Crystal structure of human butyrylcholinesterase and of its complexes with substrate and products. J. Biol. Chem. 2003, 278, 41141–41147. [Google Scholar] [CrossRef] [PubMed]
- Nachon, F.; Carletti, E.; Ronco, C.; Trovaslet, M.; Nicolet, Y.; Jean, L.; Renard, P.Y. Crystal structures of human cholinesterases in complex with huprine W and tacrine: Elements of specificity for anti-Alzheimer’s drugs targeting acetyl- and butyryl-cholinesterase. Biochem. J. 2013, 453, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Knez, D.; Brus, B.; Coquelle, N.; Sosic, I.; Sink, R.; Brazzolotto, X.; Mravljak, J.; Colletier, J.P.; Gobec, S. Structure-based development of nitroxoline derivatives as potential multifunctional anti-Alzheimer agents. Bioorganic Med. Chem. 2015, 23, 4442–4452. [Google Scholar] [CrossRef] [PubMed]
- Ellman, G.L.; Courtney, K.D.; Andres, V.J.; Feather-Stone, R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961, 7, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Dighe, S.N.; Deora, G.S.; De la Mora, E.; Nachon, F.; Chan, S.; Parat, M.O.; Brazzolotto, X.; Ross, B.P. Discovery and structure-activity relationships of a highly selective butyrylcholinesterase Inhibitor by structure-based virtual screening. J. Med. Chem. 2016, 59, 7683–7689. [Google Scholar] [CrossRef]
- Wu, C.; Cui, X.; Sun, L.; Lu, J.; Li, F.; Song, M.; Zhang, Y.; Hao, X.; Tian, C.; Song, M.; et al. Aspulvinones suppress postprandial hyperglycemia as potent α-glucosidase inhibitors from Aspergillus terreus ASM-1. Front. Chem. 2021, 9, 736070. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Hornak, V.; Abel, R.; Okur, A.; Strockbine, B.; Roitberg, A.; Simmerling, C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006, 65, 712–725. [Google Scholar] [CrossRef] [PubMed]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Ylilauri, M.; Pentikäinen, O.T. MMGBSA as a tool to understand the binding affinities of filamin-peptide interactions. J. Chem. Inf. Model. 2013, 53, 2626–2633. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 a | 2 a | 3 b | 4 b | 5 b | 6 a |
|---|---|---|---|---|---|---|
| 2 | 6.59 d (2.2) | 6.42 d (2.4) | 6.26 d (2.4) | 6.28 d (2.4) | 6.25 d (2.4) | 6.63 d (2.2) |
| 4 | 6.65 dd (8.3, 2.2) | 6.37 dd (8.2, 2.4) | 6.23 dd (8.3, 2.4) | 6.24 dd (8.3, 2.4) | 6.37 dd (8.8, 2.4) | 6.68 dd (8.4, 2.2) |
| 5 | 7.54 d (8.3) | 6.80 d (8.2) | 6.91 d (8.3) | 6.93 d (8.3) | 7.77 d (8.8) | 7.67 d (8.4) |
| 7 | — | — | 3.85 dd (8.0, 7.3) | 3.83 dd (8.2, 7.2) | — | — |
| 8 | 7.64 d (12.2) | 7.60 d (11.8) | Ha 2.59 ddd (14.6, 8.0, 7.3) Hb 2.37 dt (14.6, 7.3) | Ha 2.59 ddd (14.7, 8.2, 7.2) Hb 2.36 dt (14.7, 7.2) | 3.10–2.93 m | 7.36 d (12.6) |
| 9 | 7.39 br d (12.2) | 5.86 br d (11.8) | 5.08 br t (7.3) | 5.09 br t (7.2) | 1.96–1.76 m | 6.81 br d (12.6) |
| 11 | 2.04 br s | 1.91 br s | 1.64 d (1.4) | 1.63 d (1.7) | 1.24 s | 2.04 br s |
| 12 | 2.01 br s | 1.79 br s | 1.57 d (1.3) | 1.57 d (1.4) | 1.24 s | 2.07 br s |
| 1′ | — | 3.64 s | 3.62 s | 4.09 dq (16.6, 7.1) 4.07 dq (16.6, 7.1) | — | — |
| 2′ | — | — | — | 1.18 t (7.1) | — | — |
| 1-OH | — | 7.97 br s | — | — | — | — |
| 3-OH | 8.93 br s | 8.26 br s | — | — | — | 8.94 br s |
| No. | 1 b | 2 b | 3 c | 4 c | 5 c | 6 b |
|---|---|---|---|---|---|---|
| 1 | 154.9 s | 157.0 s | 157.0 s | 156.9 s | 166.4 s | 155.9 s |
| 2 | 98.9 d | 103.6 d | 103.4 d | 103.4 d | 103.7 d | 99.2 d |
| 3 | 160.0 s | 159.2 s | 158.3 s | 158.2 s | 166.5 s | 160.1 s |
| 4 | 111.9 d | 107.3 d | 107.5 d | 107.4 d | 109.1 d | 112.1 d |
| 5 | 121.9 d | 133.1 d | 129.9 d | 129.8 d | 133.7 d | 125.4 d |
| 6 | 117.5 s | 115.2 s | 118.3 s | 118.4 s | 113.9 s | 115.9 s |
| 7 | 117.9 s | 128.4 s | 45.6 d | 45.8 d | 206.5 s | 118.7 s |
| 8 | 133.2 d | 136.8 d | 32.1 t | 32.1 t | 34.0 t | 132.2 d |
| 9 | 122.3 d | 123.2 d | 123.0 d | 122.9 d | 39.1 t | 122.0 d |
| 10 | 151.0 s | 145.3 s | 134.2 s | 134.1 s | 70.9 s | 153.0 s |
| 11 | 18.7 q | 18.9 q | 25.9 q | 25.9 q | 29.2 q | 18.9 q |
| 12 | 27.3 q | 26.8 q | 17.8 q | 17.8 q | 29.2 q | 27.2 q |
| 13 | 167.8 s | 169.1 s | 177.3 s | 176.9 s | — | 170.1 s |
| 1′ | — | 51.8 q | 52.1 q | 61.5 t | — | — |
| 2′ | — | — | — | 14.5 q | — | — |
| Compounds | IC50 μM a | BChE | ||||
|---|---|---|---|---|---|---|
| AChE | BChE | Ki μM | αKi μM | α | Inhibition Type | |
| 1 | >100 | 45.5 ± 5.9 | 93.4 | 43.0 | 0.46 | Mixed uncompetitive |
| 2 | >100 | >100 | — | — | — | — |
| 3 | >100 | >100 | — | — | — | — |
| 4 | >100 | 61.0 ± 3.4 | — | — | — | — |
| 5 | >100 | >100 | — | — | — | — |
| 6 | >100 | >100 | — | — | — | — |
| 7 | >100 | >100 | — | — | — | — |
| 8 | >100 | 27.9 ± 3.5 | 12.8 | 71.1 | 5.55 | Mixed competitive |
| 9 | 81.2 ± 3.9 | 9.5 ± 3.2 | 5.7 | 25.2 | 4.42 | Mixed competitive |
| 10 | >100 | >100 | — | — | — | — |
| 11 | 40.5 ± 4.7 | 13.5 ± 1.6 | 12.7 | 13.3 | 1.05 | Noncompetitive |
| 14 | >100 | 2.6 ± 0.2 | 1.7 | 22.7 | 13.35 | Competitive |
| 15 | >100 | 27.1 ± 6.8 | — | — | — | — |
| 16 | >100 | 32.8 ± 4.2 | — | — | — | — |
| Galantamine b | 0.8 ± 0.2 | 35.3 ± 5.6 | — | — | — | — |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cui, X.; Huang, Z.; Deng, S.; Zhang, Y.; Li, G.; Wang, L.; Deng, Y.; Wu, C. Benzofuran Derivatives from Cortex Mori Radicis and Their Cholinesterase-Inhibitory Activity. Molecules 2024, 29, 315. https://doi.org/10.3390/molecules29020315
Cui X, Huang Z, Deng S, Zhang Y, Li G, Wang L, Deng Y, Wu C. Benzofuran Derivatives from Cortex Mori Radicis and Their Cholinesterase-Inhibitory Activity. Molecules. 2024; 29(2):315. https://doi.org/10.3390/molecules29020315
Chicago/Turabian StyleCui, Xiang, Zehong Huang, Shanshan Deng, Yunxia Zhang, Guoyin Li, Lining Wang, Yanru Deng, and Changjing Wu. 2024. "Benzofuran Derivatives from Cortex Mori Radicis and Their Cholinesterase-Inhibitory Activity" Molecules 29, no. 2: 315. https://doi.org/10.3390/molecules29020315
APA StyleCui, X., Huang, Z., Deng, S., Zhang, Y., Li, G., Wang, L., Deng, Y., & Wu, C. (2024). Benzofuran Derivatives from Cortex Mori Radicis and Their Cholinesterase-Inhibitory Activity. Molecules, 29(2), 315. https://doi.org/10.3390/molecules29020315