
Electronic Properties of Graphene Nano-Parallelograms: A Thermally Assisted Occupation DFT Computational Study



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
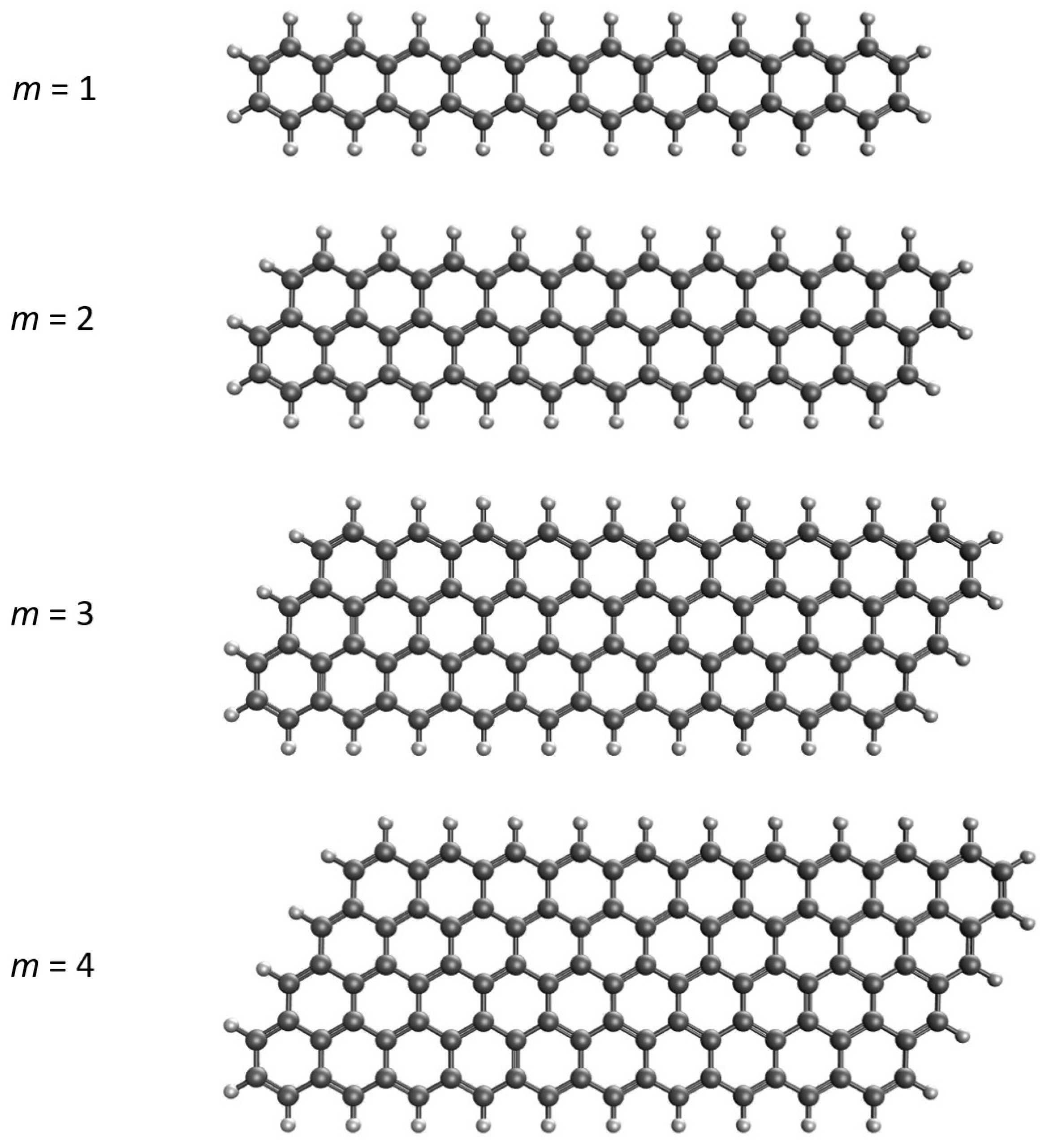
:1. Introduction
2. Results and Discussion
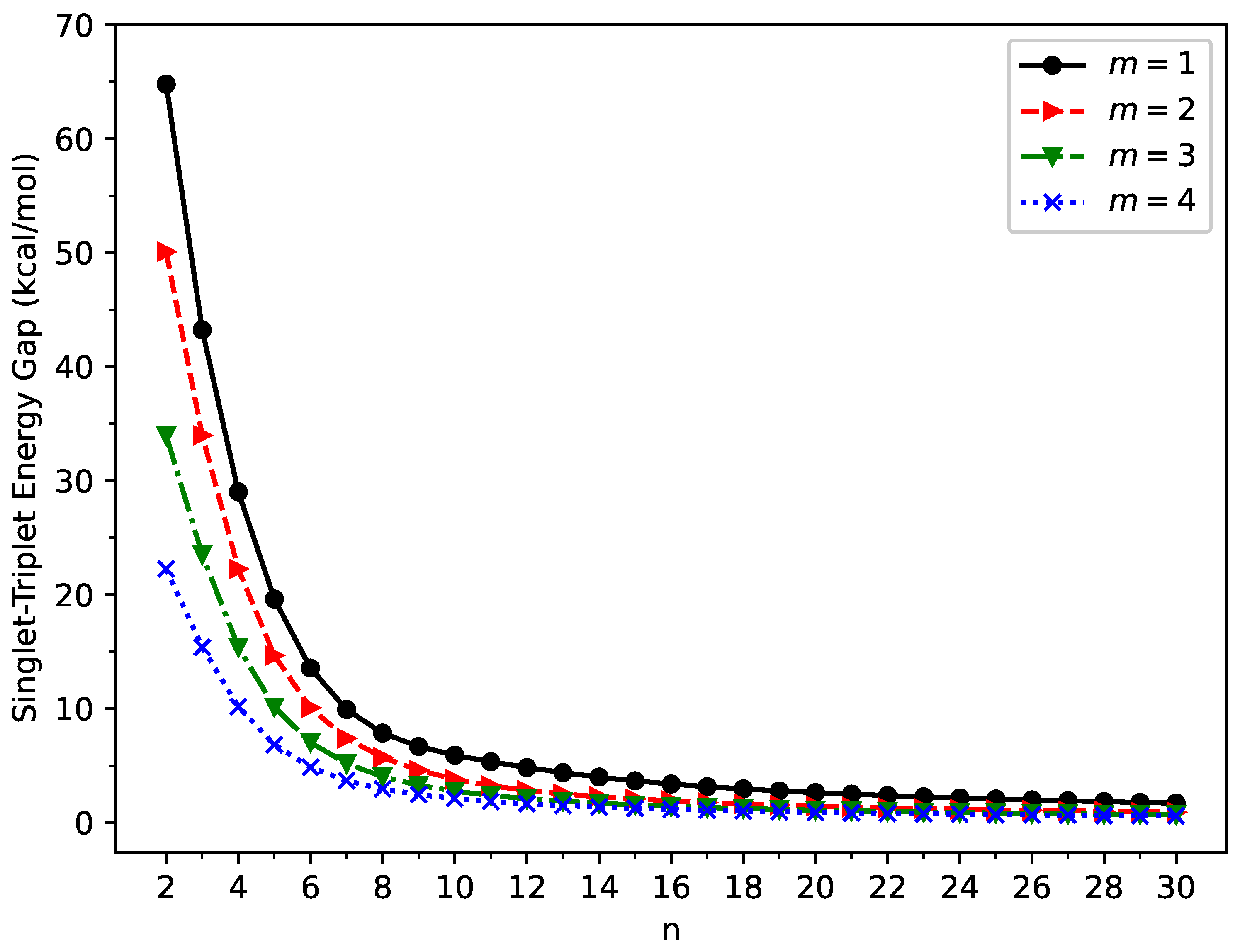
2.1. Singlet–Triplet Energy Gap
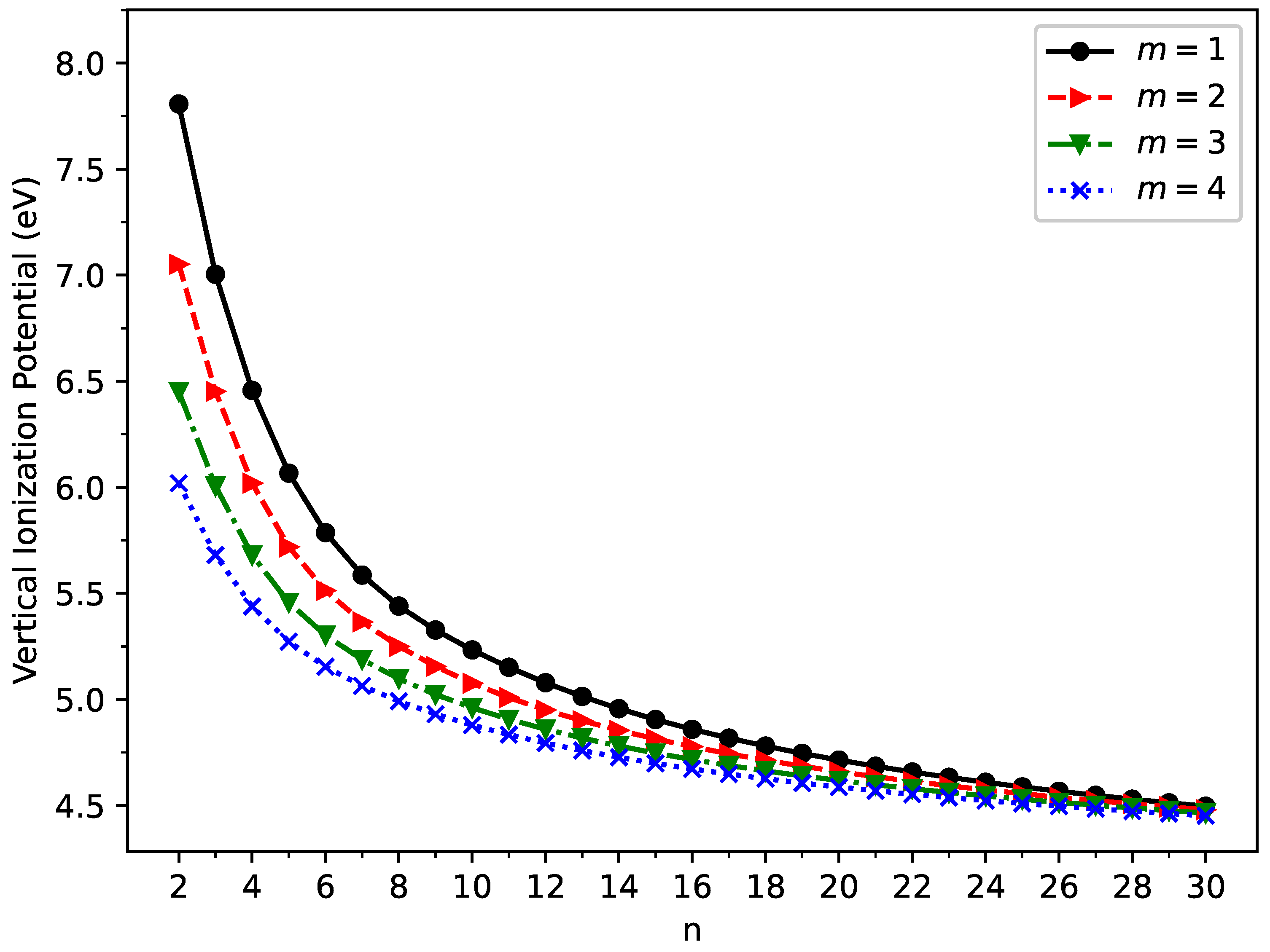
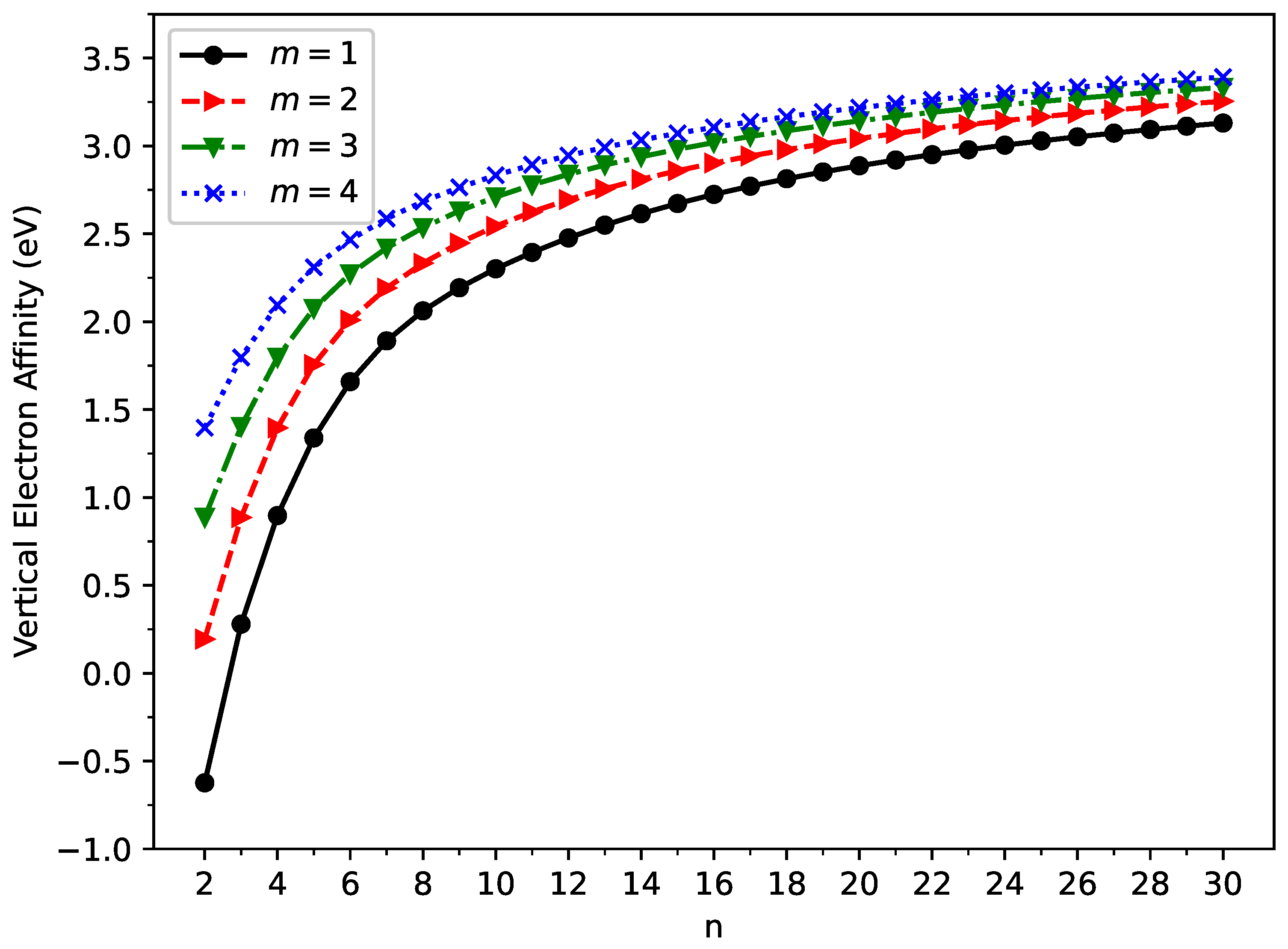
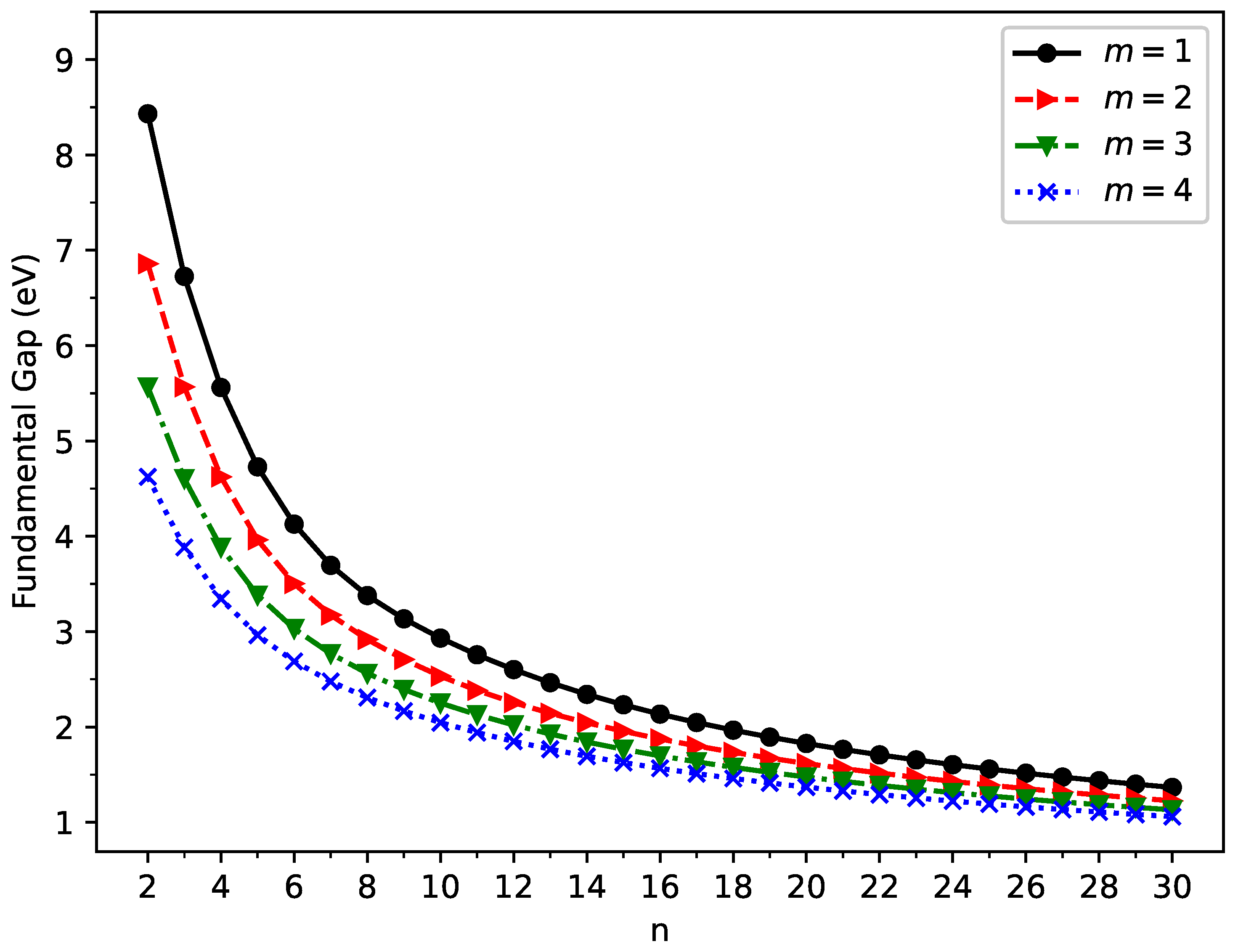
2.2. Vertical Ionization Potential, Vertical Electron Affinity, and Fundamental Gap
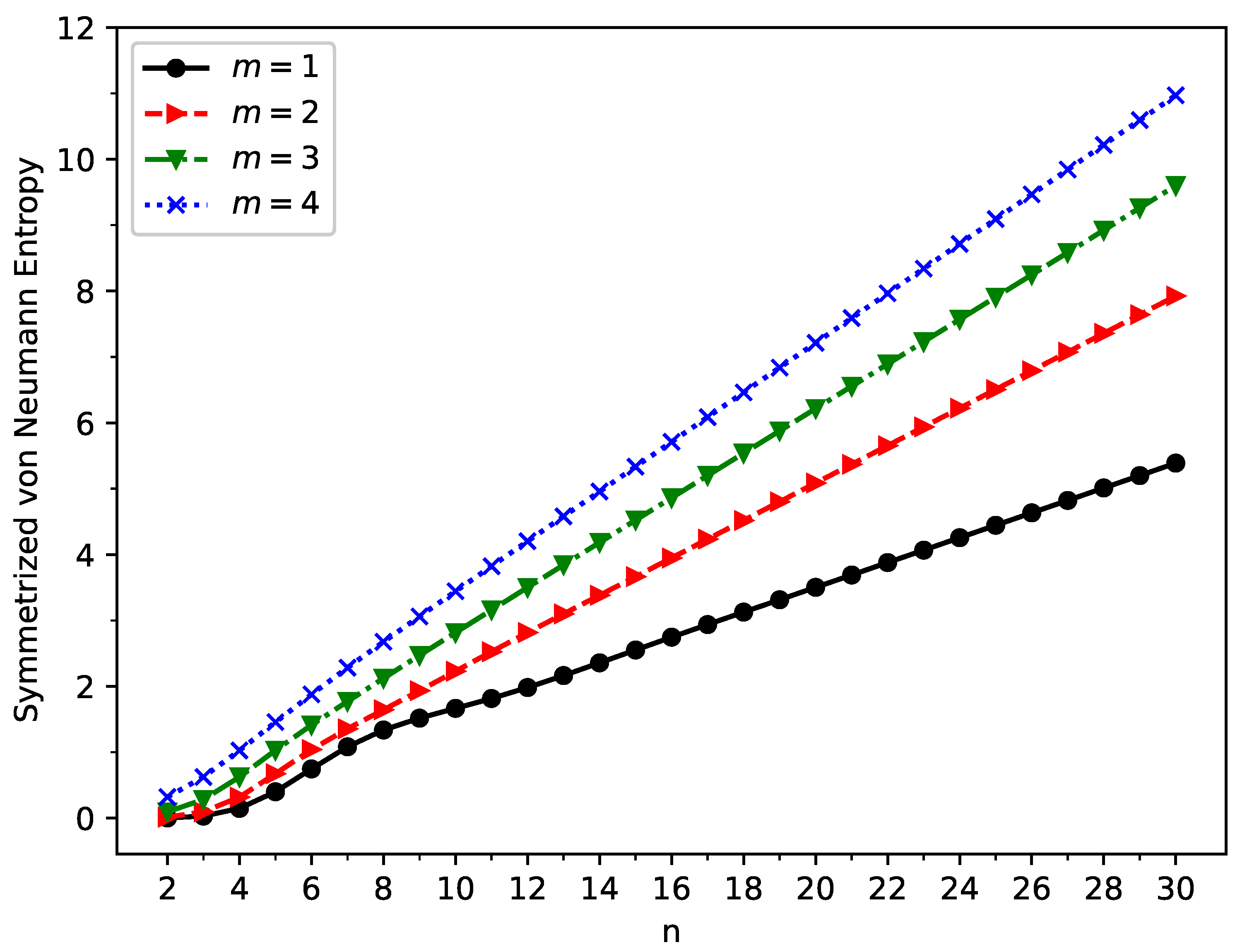
2.3. Symmetrized von Neumann Entropy
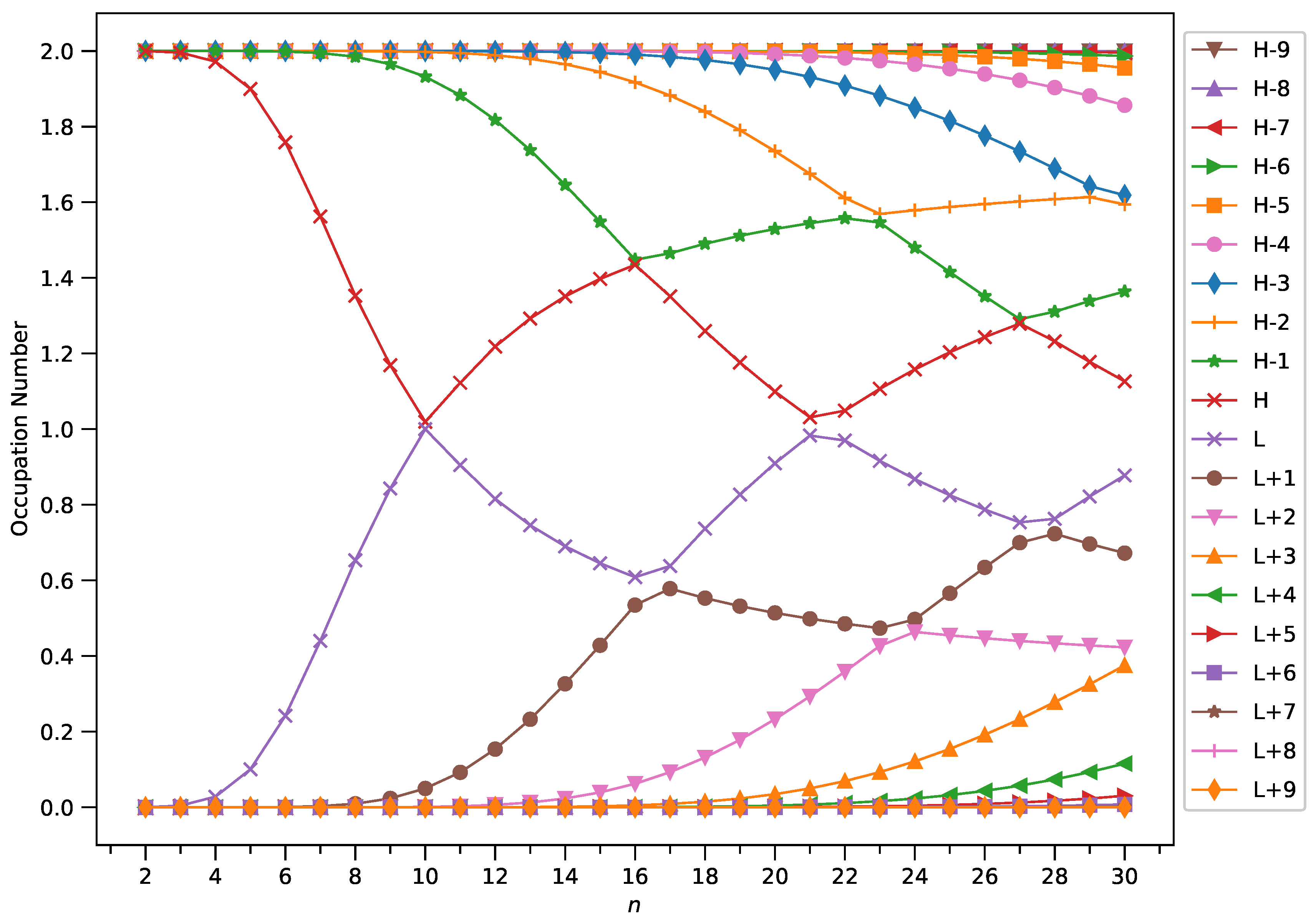
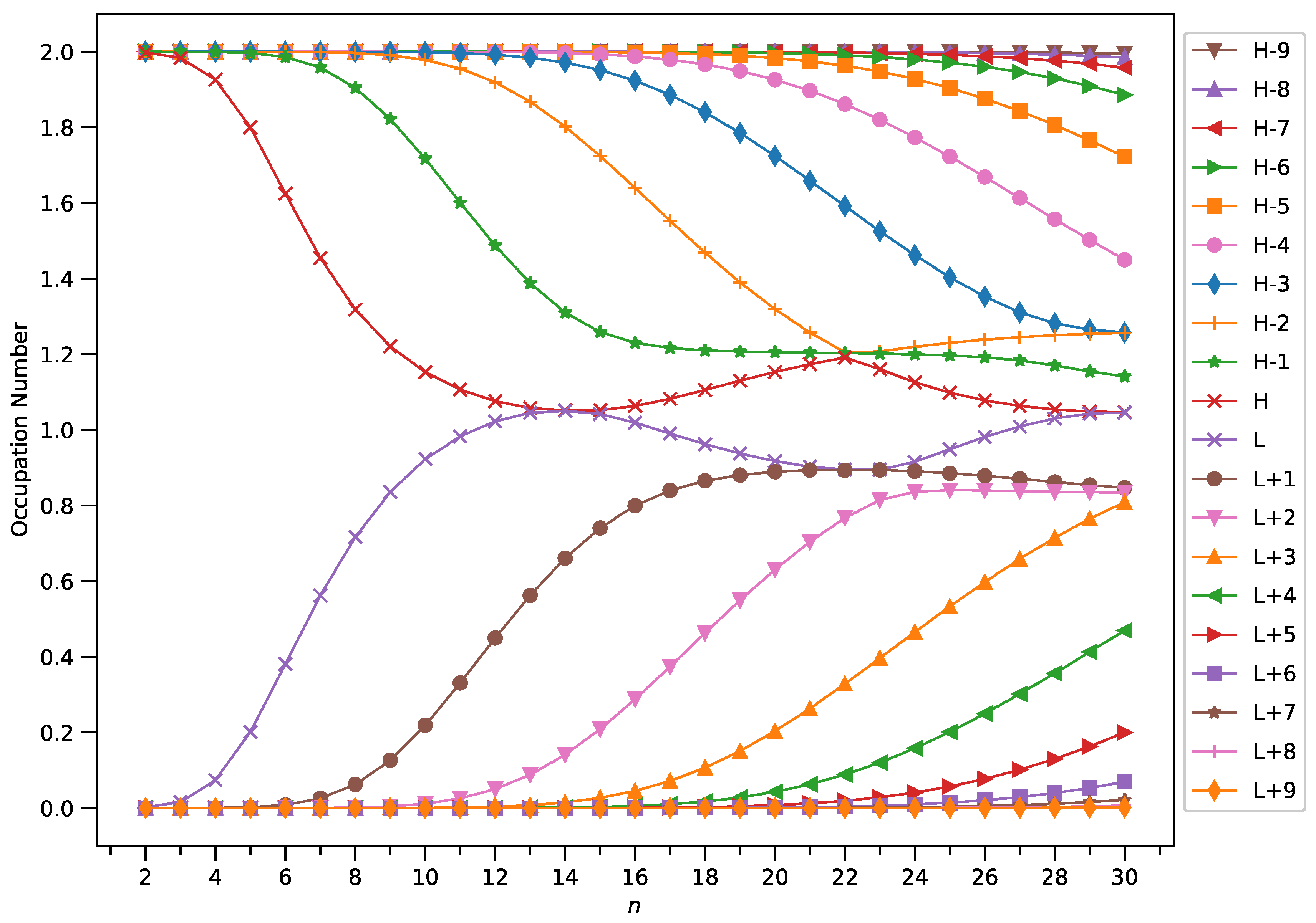
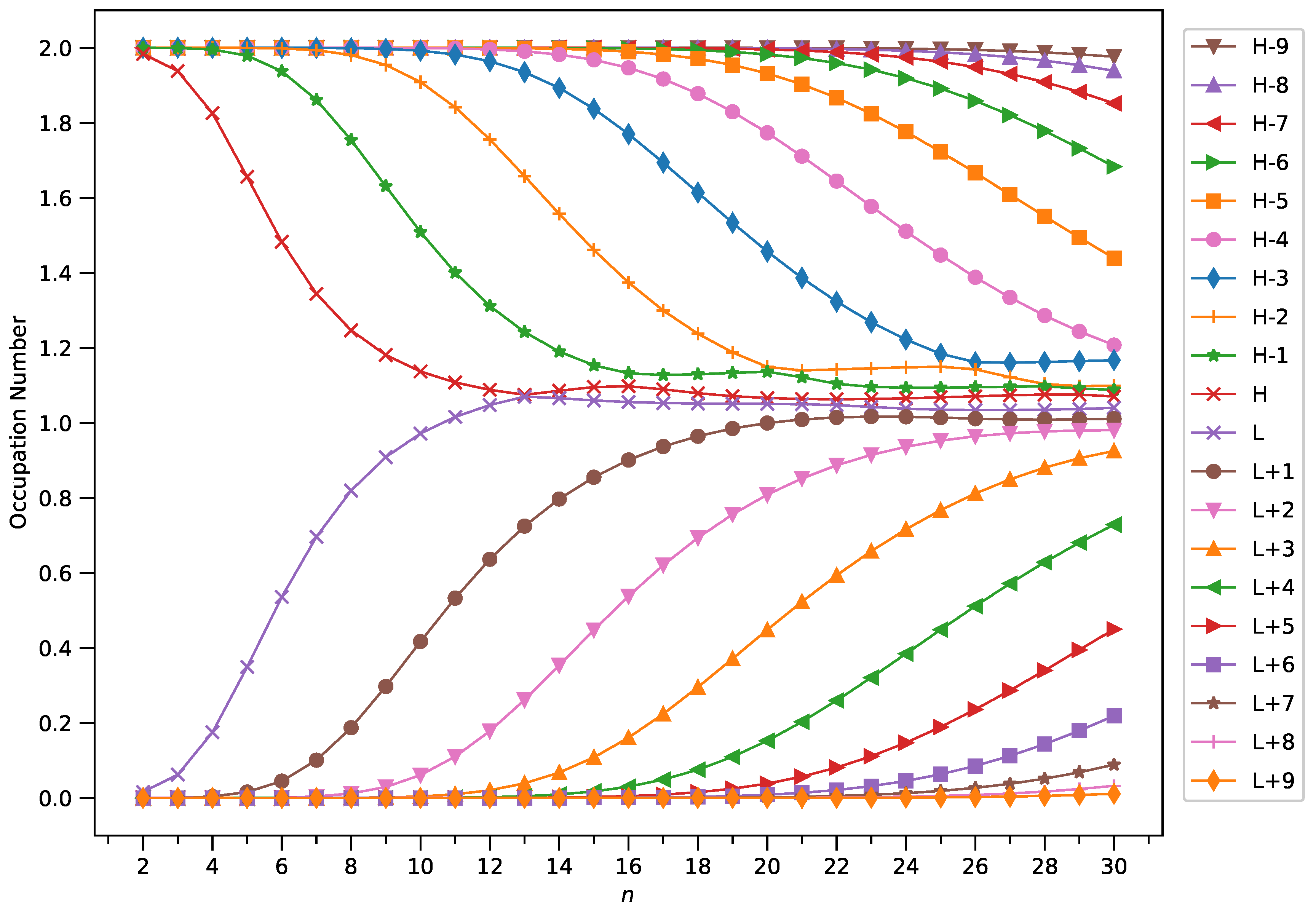
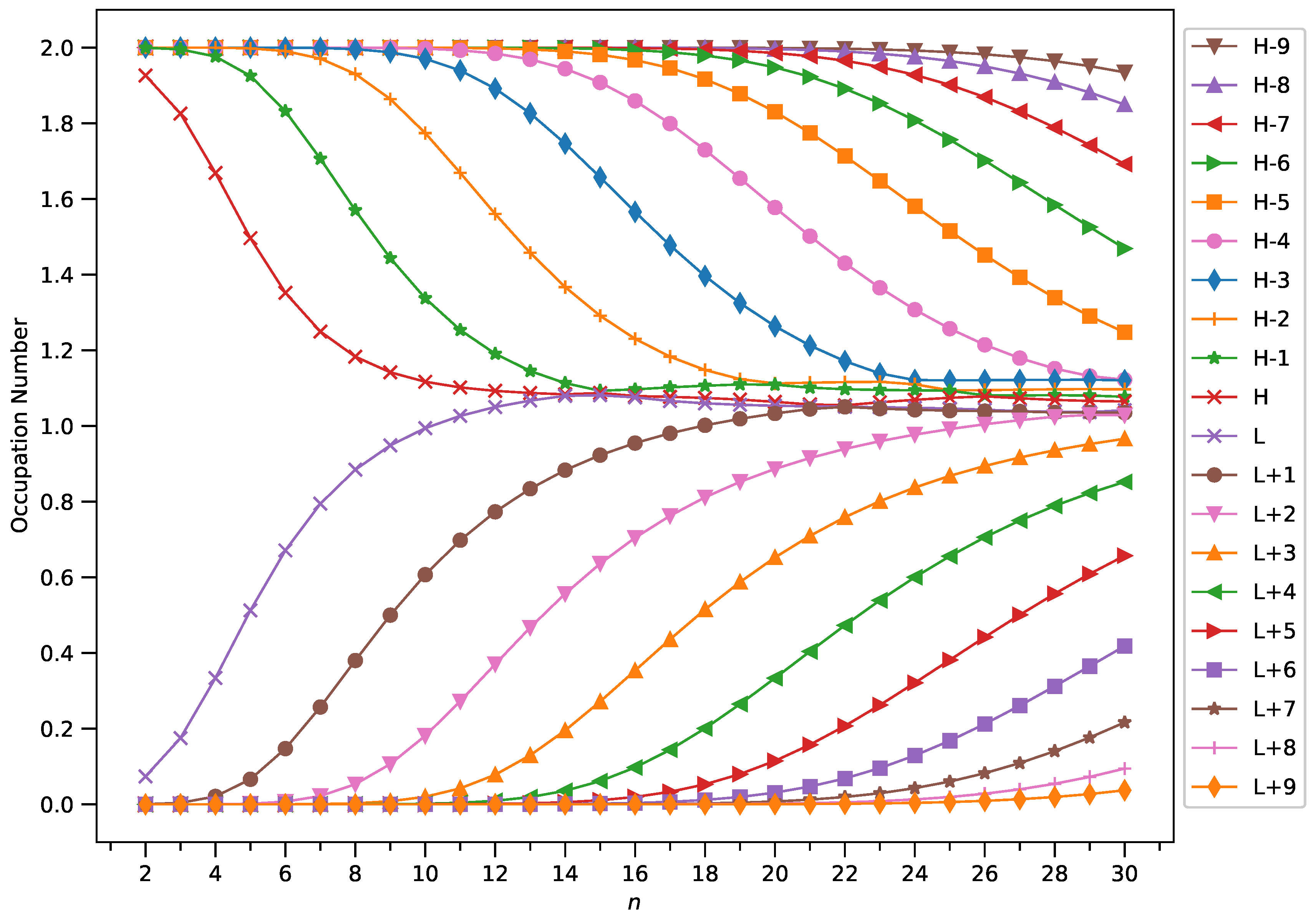
2.4. Active Orbital Occupation Numbers
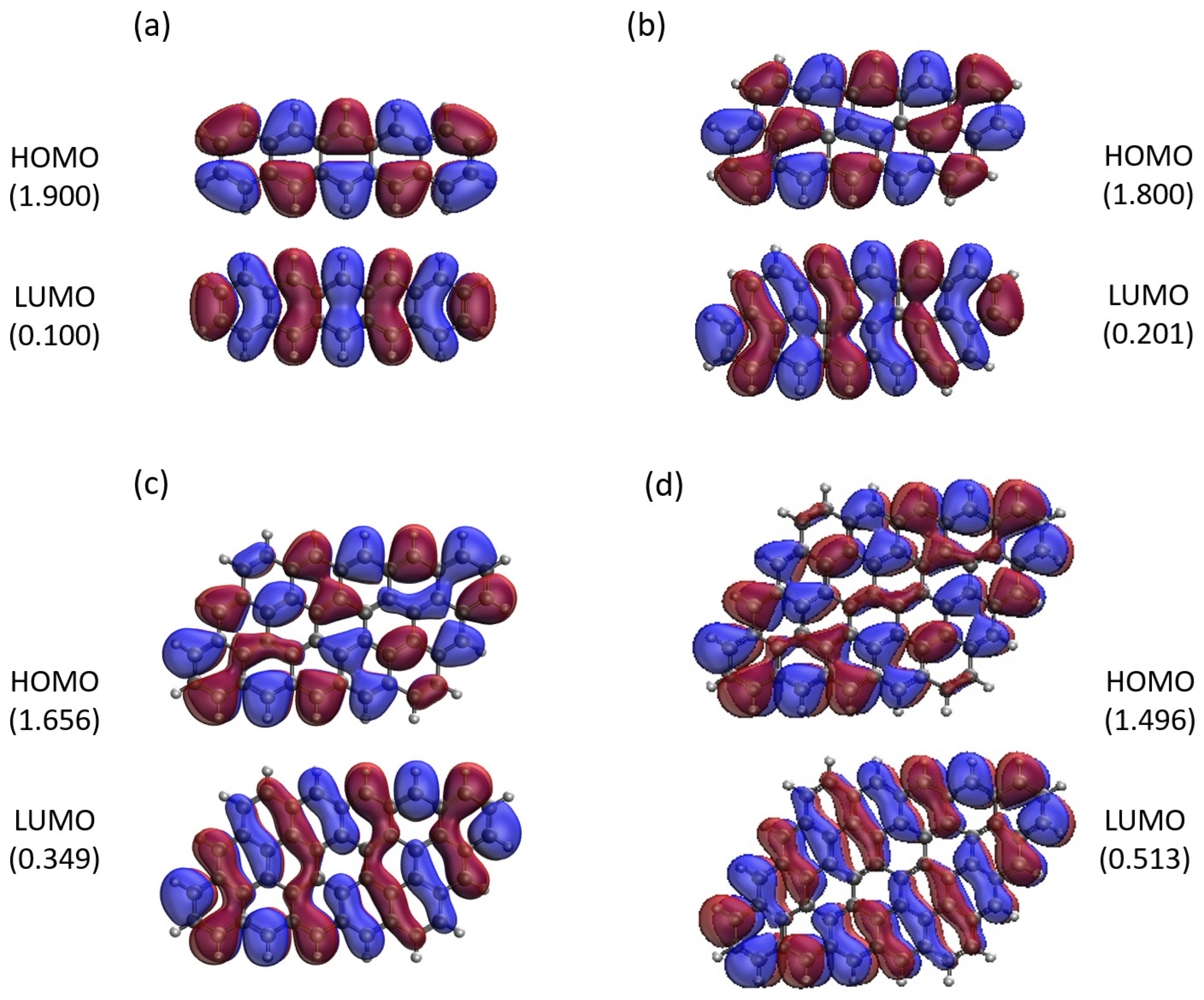
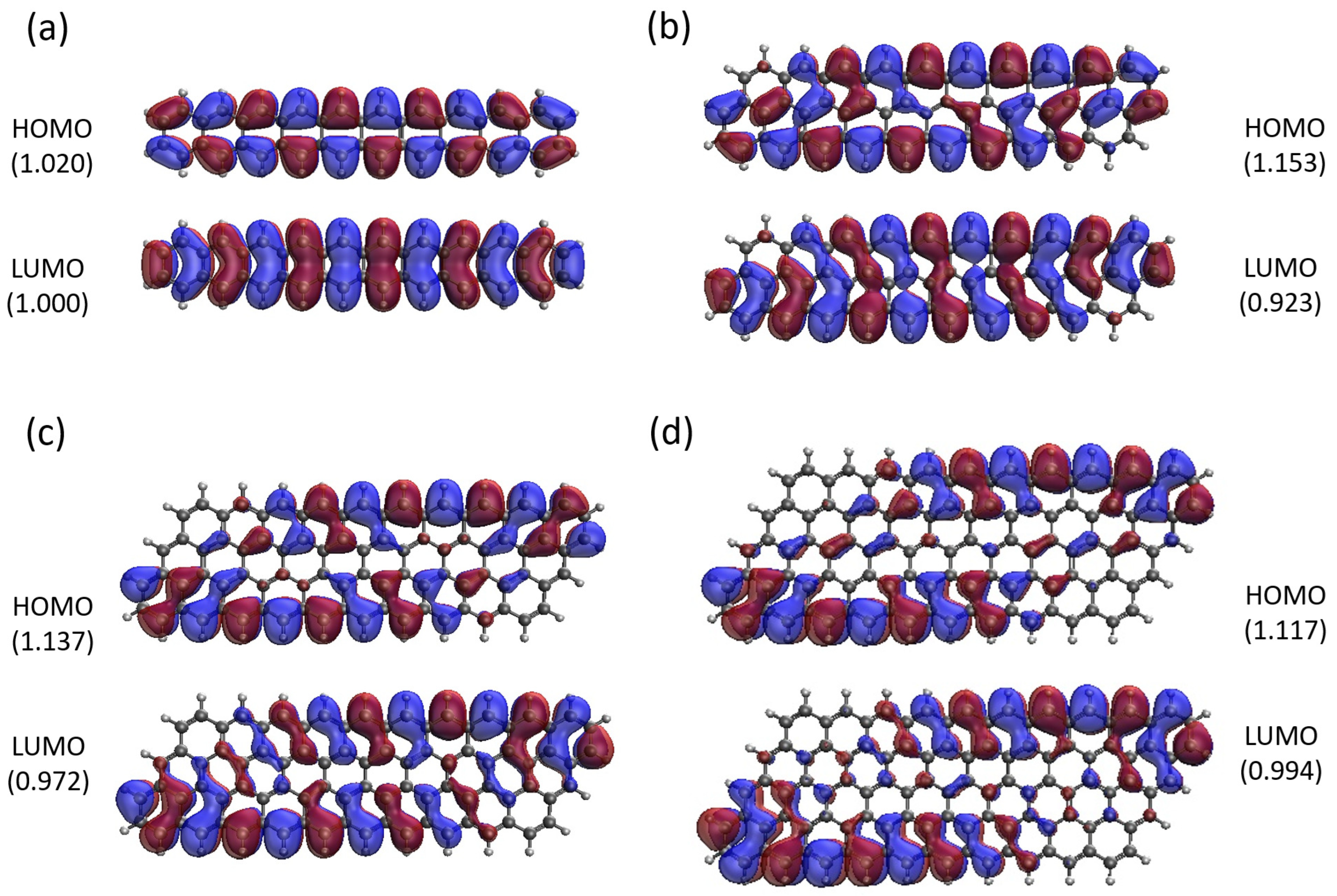
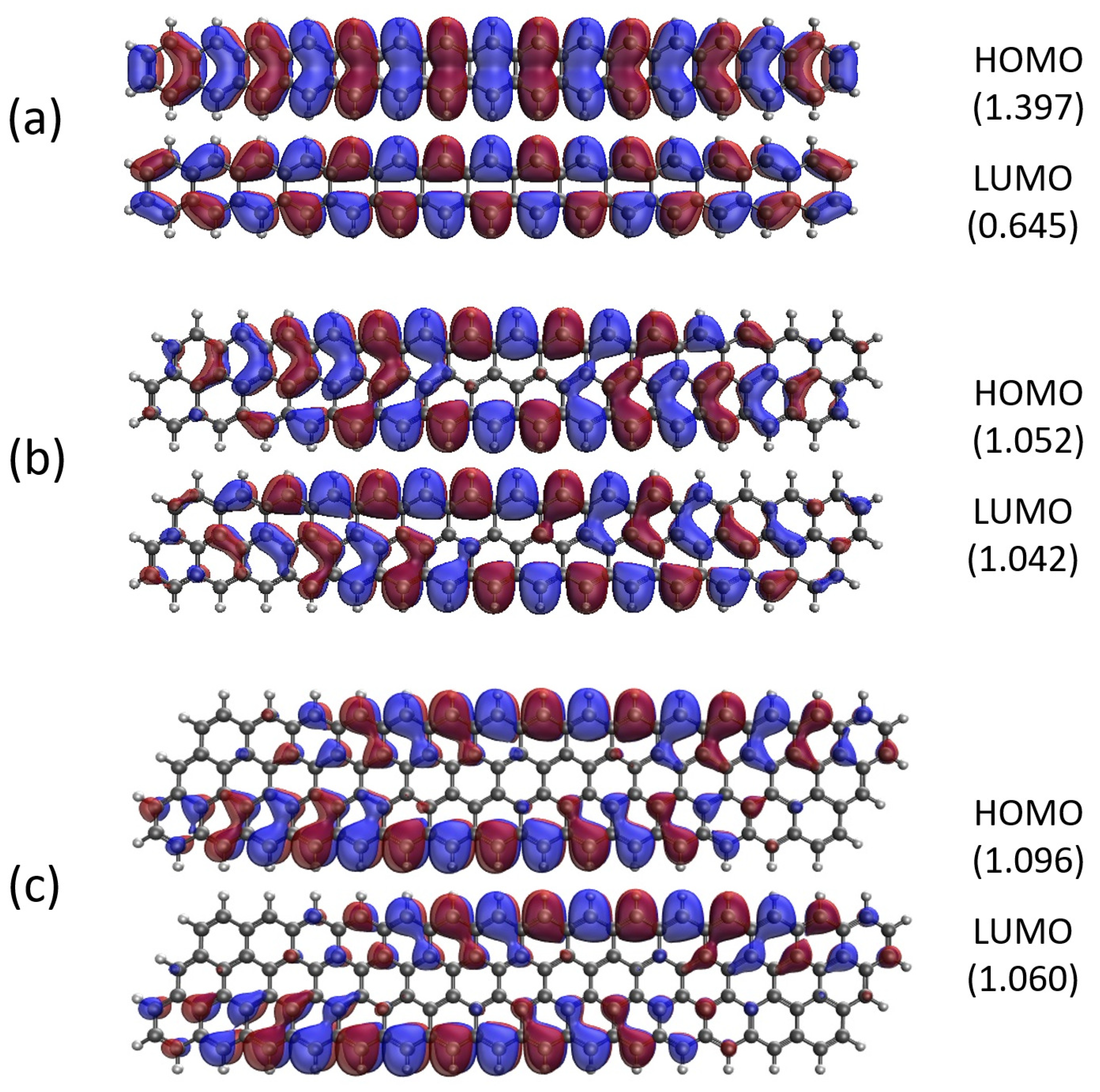
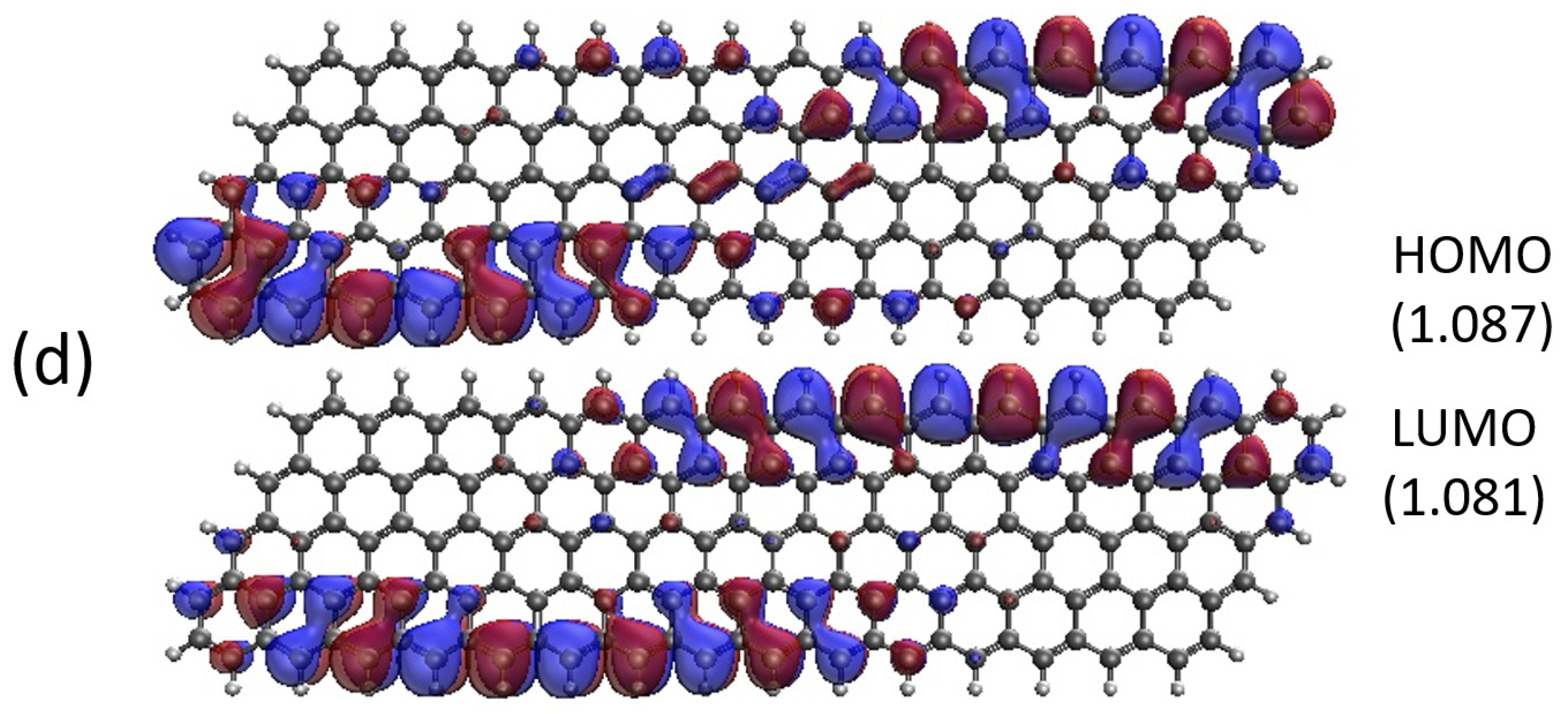
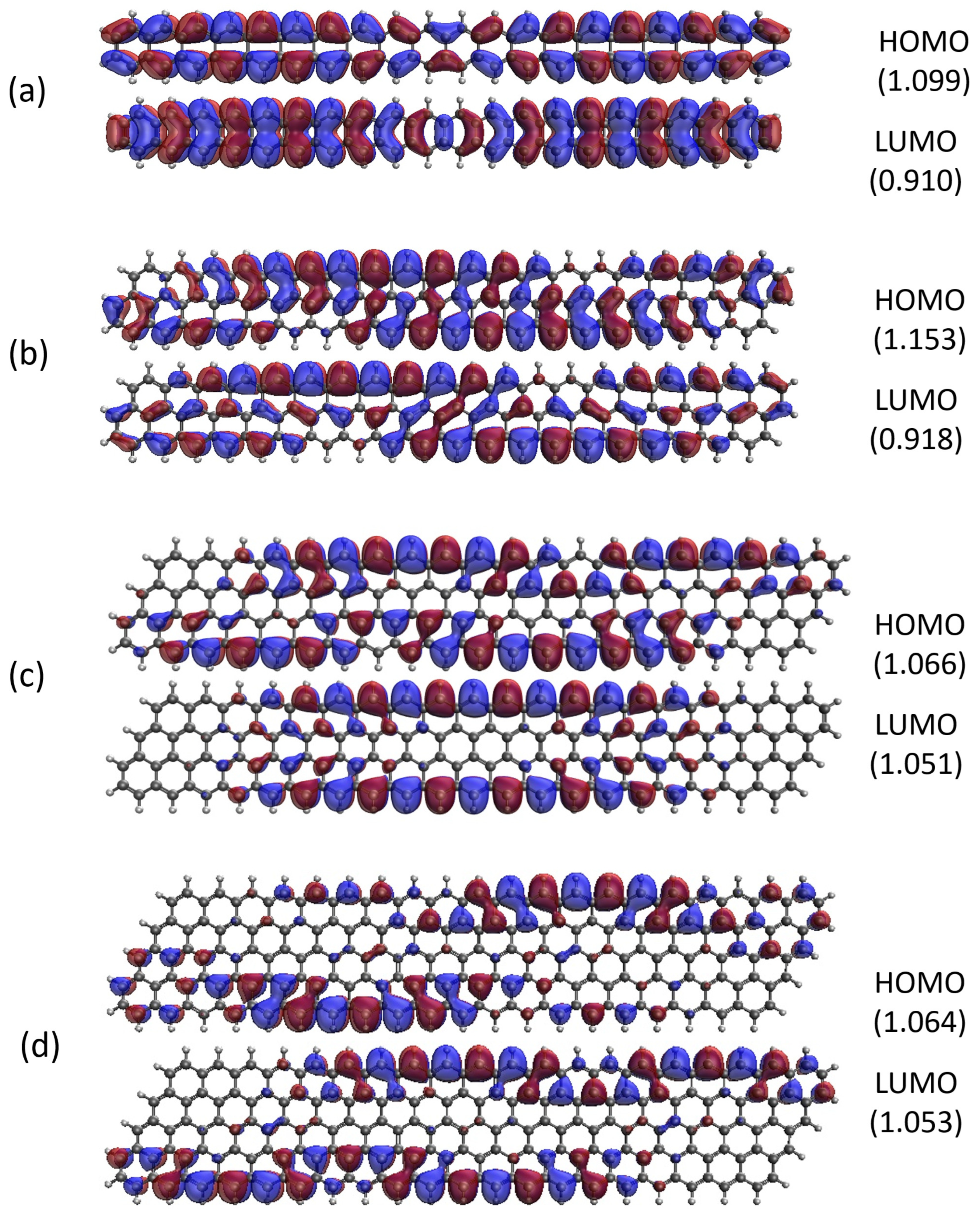
2.5. Real-Space Representation of Active Orbitals
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hirsch, A. The era of carbon allotropes. Nat. Mater. 2010, 9, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Georgakilas, V.; Perman, J.A.; Tucek, J.; Zboril, R. Broad family of carbon nanoallotropes: Classification, chemistry, and applications of fullerenes, carbon dots, nanotubes, graphene, nanodiamonds, and combined superstructures. Chem. Rev. 2015, 115, 4744–4822. [Google Scholar] [CrossRef] [PubMed]
- Kroto, H.W.; Heath, J.R.; O’Brien, S.C.; Curl, R.F.; Smalley, R.E. C60: Buckminsterfullerene. Nature 1985, 318, 162–163. [Google Scholar] [CrossRef]
- Meirzadeh, E.; Evans, A.M.; Rezaee, M.; Milich, M.; Dionne, C.J.; Darlington, T.P.; Bao, S.T.; Bartholomew, A.K.; Handa, T.; Rizzo, D.J.; et al. A few-layer covalent network of fullerenes. Nature 2023, 613, 71–76. [Google Scholar] [CrossRef]
- Iijima, S. Helical microtubules of graphitic carbon. Nature 1991, 354, 56–58. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Geim, A.K.; Morozov, S.V.; Jiang, D.E.; Zhang, Y.; Dubonos, S.V.; Grigorieva, I.V.; Firsov, A.A. Electric field effect in atomically thin carbon films. Science 2004, 306, 666–669. [Google Scholar] [CrossRef] [PubMed]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef]
- Madurani, K.A.; Suprapto, S.; Machrita, N.I.; Bahar, S.L.; Illiya, W.; Kurniawan, F. Progress in graphene synthesis and its application: History, challenge and the future outlook for research and industry. ECS J. Solid State Sci. Technol. 2020, 9, 093013. [Google Scholar] [CrossRef]
- Houtsma, R.K.; de la Rie, J.; Stöhr, M. Atomically precise graphene nanoribbons: Interplay of structural and electronic properties. Chem. Soc. Rev. 2021, 50, 6541–6568. [Google Scholar] [CrossRef]
- Gu, Y.; Qiu, Z.; Müllen, K. Nanographenes and graphene nanoribbons as multitalents of present and future materials science. J. Am. Chem. Soc. 2022, 144, 11499–11524. [Google Scholar] [CrossRef]
- Han, M.Y.; Özyilmaz, B.; Zhang, Y.; Kim, P. Energy band-gap engineering of graphene nanoribbons. Phys. Rev. Lett. 2007, 98, 206805. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, N.; Menchón, R.E.; Pozo, I.; Hieulle, J.; Vegliante, A.; Li, J.; Sánchez-Portal, D.; Penã, D.; Garcia-Lekue, A.; Pascual, J.I. Addressing electron spins embedded in metallic graphene nanoribbons. ACS Nano 2022, 16, 14819–14826. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.W.; Cohen, M.L.; Louie, S.G. Energy gaps in graphene nanoribbons. Phys. Rev. Lett. 2006, 97, 216803. [Google Scholar] [CrossRef] [PubMed]
- Kimouche, A.; Ervasti, M.M.; Drost, R.; Halonen, S.; Harju, A.; Joensuu, P.M.; Sainio, J.; Liljeroth, P. Ultra-narrow metallic armchair graphene nanoribbons. Nat. Commun. 2015, 6, 10177. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Neuman, T.; Boeglin, A.; Scheurer, F.; Schull, G. Topologically localized excitons in single graphene nanoribbons. Science 2023, 379, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Pratap, S.; Kumar, V.; Mishra, R.K.; Gwag, J.S.; Chakraborty, B. Electronic, transport, magnetic, and optical properties of graphene nanoribbons and their optical sensing applications: A comprehensive review. Luminescence 2023, 38, 909–953. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, H.S.; Ma, C.; Chen, L.; Jiang, C.; Chen, C.; Xie, X.; Li, A.P.; Wang, X. Graphene nanoribbons for quantum electronics. Nat. Rev. Phys. 2021, 3, 791–802. [Google Scholar] [CrossRef]
- Saraswat, V.; Jacobberger, R.M.; Arnold, M.S. Materials science challenges to graphene nanoribbon electronics. ACS Nano 2021, 15, 3674–3708. [Google Scholar] [CrossRef]
- Luo, H.; Yu, G. Preparation, bandgap engineering, and performance control of graphene nanoribbons. Chem. Mater. 2022, 34, 3588–3615. [Google Scholar] [CrossRef]
- Yano, Y.; Mitoma, N.; Ito, H.; Itami, K. A quest for structurally uniform graphene nanoribbons: Synthesis, properties, and applications. J. Org. Chem. 2019, 85, 4–33. [Google Scholar] [CrossRef]
- Gu, Y.; Wu, X.; Gopalakrishna, T.Y.; Phan, H.; Wu, J. Graphene-like molecules with four zigzag edges. Ang. Chem. Int. Ed. 2018, 57, 6541–6545. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Tullimilli, Y.G.; Feng, J.; Phan, H.; Zeng, W.; Wu, J. peri-Acenoacenes. Chem. Comm. 2019, 55, 5567–5570. [Google Scholar] [CrossRef]
- Muñoz–Mármol, R.; Gordillo, F.; Bonal, V.; Villalvilla, J.M.; Boj, P.G.; Quintana, J.A.; Ross, A.M.; Paternò, G.M.; Scotognella, F.; Lanzani, G.; et al. Near-Infrared lasing in four-zigzag edged nanographenes by 1D versus 2D electronic π-conjugation. Adv. Funct. Mater. 2021, 31, 2105073. [Google Scholar] [CrossRef]
- Jousselin-Oba, T.; Mamada, M.; Wright, K.; Marrot, J.; Adachi, C.; Yassar, A.; Frigoli, M. Synthesis, aromaticity, and application of peri-pentacenopentacene: Localized representation of benzenoid aromatic compounds. Angew. Chem. Int. Ed. 2022, 134, e202112794. [Google Scholar] [CrossRef]
- Gu, Y.; Muñoz-Mármol, R.; Fan, W.; Han, Y.; Wu, S.; Li, Z.; Bonal, V.; Villalvilla, J.M.; Quintana, J.A.; Boj, P.G.; et al. Peri-acenoacene for solution processed distributed feedback laser: The effect of 1,2-oxaborine doping. Adv. Opt. Mater. 2022, 10, 2102782. [Google Scholar] [CrossRef]
- Omist, A.; Ricci, G.; Derradji, A.; Pérez-Jiménez, A.J.; San-Fabián, E.; Olivier, Y.; Sancho-Garcia, J.C. Peri-acenoacene molecules: Tuning of the singlet and triplet excitation energies by modifying their radical character. Phys. Chem. Chem. Phys. 2021, 23, 24016–24028. [Google Scholar] [CrossRef] [PubMed]
- Sandoval-Salinas, M.E.; Bernabeu-Cabañero, R.; Pérez-Jiménez, A.J.; San-Fabián, E.; Sancho-García, J.C. Electronic structure of rhombus-shaped nanographenes: System size evolution from closed- to open-shell ground states. Phys. Chem. Chem. Phys. 2023, 25, 11697–11706. [Google Scholar] [CrossRef]
- Hauwali, N.U.J.; Syuhada, I.; Rosikhin, A.; Winata, T. Fundamental properties of parallelogram graphene nanoflakes: A first principle study. Mat. Today Proc. 2021, 44, 3305–3308. [Google Scholar] [CrossRef]
- Wu, C.-S.; Chai, J.-D. Electronic properties of zigzag graphene nanoribbons studied by TAO-DFT. J. Chem. Theory Comput. 2015, 11, 2003–2011. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, B864–B871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Teale, A.M.; Helgaker, T.; Savin, A.; Adamo, C.; Aradi, B.; Arbuznikov, A.V.; Ayers, P.W.; Baerends, E.J.; Barone, V.; Calaminici, P.; et al. DFT Exchange: Sharing perspectives on the workhorse of quantum chemistry and materials science. Phys. Chem. Chem. Phys. 2022, 24, 28700–28781. [Google Scholar] [CrossRef] [PubMed]
- Dirac, P.A.M. Note on exchange phenomena in the Thomas atom. Proc. Camb. Philos. Soc. 1930, 26, 376–385. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244–13249. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Kümmel, S.; Kronik, L. Orbital-dependent density functionals: Theory and applications. Rev. Mod. Phys. 2008, 80, 3–60. [Google Scholar] [CrossRef]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Insights into current limitations of density functional theory. Science 2008, 321, 792–794. [Google Scholar] [CrossRef]
- Engel, E.; Dreizler, R.M. Density Functional Theory: An Advanced Course; Springer: Heidelberg, Germany, 2011. [Google Scholar]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Challenges for density functional theory. Chem. Rev. 2012, 112, 289–320. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Grimme, S.; Hansen, A.; Brandenburg, J.G.; Bannwarth, C. Dispersion-corrected mean-field electronic structure methods. Chem. Rev. 2016, 116, 5105–5154. [Google Scholar] [CrossRef] [PubMed]
- Andersson, K.; Malmqvist, P.-Å.; Roos, B.O. Second-order perturbation theory with a complete active space self-consistent field reference function. J. Chem. Phys. 1992, 96, 1218. [Google Scholar] [CrossRef]
- Hachmann, J.; Dorando, J.J.; Aviles, M.; Chan, G.K.L. The radical character of the acenes: A density matrix renormalization group study. J. Chem. Phys. 2007, 127, 134309. [Google Scholar] [CrossRef]
- Gidofalvi, G.; Mazziotti, D.A. Active-space two-electron reduced-density-matrix method: Complete active-space calculations without diagonalization of the N-electron hamiltonian. J. Chem. Phys. 2008, 129, 134108. [Google Scholar] [CrossRef] [PubMed]
- Mizukami, W.; Kurashige, Y.; Yanai, T. More π electrons make a difference: Emergence of many radicals on graphene nanoribbons studied by ab initio DMRG theory. J. Chem. Theory Comput. 2013, 9, 401–407. [Google Scholar] [CrossRef]
- Gryn’ova, G.; Coote, M.L.; Corminboeuf, C. Theory and practice of uncommon molecular electronic configurations. WIREs Comput. Mol. Sci. 2015, 5, 440–459. [Google Scholar] [CrossRef]
- Fosso-Tande, J.; Nguyen, T.-S.; Gidofalvi, G.; DePrince III, A.E. Large-scale variational two-electron reduced-density-matrix-driven complete active space self-consistent field methods. J. Chem. Theory Comput. 2016, 12, 2260–2271. [Google Scholar] [CrossRef]
- Piris, M. Global method for electron correlation. Phys. Rev. Lett. 2017, 119, 063002. [Google Scholar] [CrossRef]
- Goli, V.D.P.; Prodhan, S.; Mazumdar, S.; Ramasesha, S. Correlated electronic properties of some graphene nanoribbons: A DMRG study. Phys. Rev. B 2016, 94, 035139. [Google Scholar] [CrossRef]
- Hagymási, I.; Legeza, Ö. Entanglement, excitations, and correlation effects in narrow zigzag graphene nanoribbons. Phys. Rev. B 2016, 94, 165147. [Google Scholar] [CrossRef]
- Chai, J.-D. Density functional theory with fractional orbital occupations. J. Chem. Phys. 2012, 136, 154104. [Google Scholar] [CrossRef]
- Chai, J.-D. Thermally-assisted-occupation density functional theory with generalized-gradient approximations. J. Chem. Phys. 2014, 140, 18A521. [Google Scholar] [CrossRef]
- Chai, J.-D. Role of exact exchange in thermally-assisted-occupation density functional theory: A proposal of new hybrid schemes. J. Chem. Phys. 2017, 146, 044102. [Google Scholar] [CrossRef] [PubMed]
- Xuan, F.; Chai, J.-D.; Su, H. Local density approximation for the short-range exchange free energy functional. ACS Omega 2019, 4, 7675–7683. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.-Y.; Chai, J.-D. Real-time extension of TAO-DFT. Molecules 2023, 28, 7247. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.-J.; Chai, J.-D. TAO-DFT fictitious temperature made simple. RSC Adv. 2022, 12, 12193–12210. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Hui, K.; Chung, J.-H.; Chai, J.-D. Self-consistent determination of the fictitious temperature in thermally-assisted-occupation density functional theory. RSC Adv. 2017, 7, 50496–50507. [Google Scholar] [CrossRef]
- Mermin, N.D. Thermal properties of the inhomogeneous electron gas. Phys. Rev. 1965, 137, A1441–A1443. [Google Scholar] [CrossRef]
- Li, S.; Chai, J.-D. TAO-DFT-based ab initio molecular dynamics. Front. Chem. 2020, 8, 589432. [Google Scholar] [CrossRef] [PubMed]
- Seenithurai, S.; Chai, J.-D. TAO-DFT with the polarizable continuum model. Nanomaterials 2023, 13, 1593. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.-N.; Chai, J.-D. Role of Kekulé and non-Kekulé structures in the radical character of alternant polycyclic aromatic hydrocarbons: A TAO-DFT study. Sci. Rep. 2016, 6, 30562. [Google Scholar] [CrossRef] [PubMed]
- Seenithurai, S.; Chai, J.-D. Effect of Li adsorption on the electronic and hydrogen storage properties of acenes: A dispersion-corrected TAO-DFT study. Sci. Rep. 2016, 6, 33081. [Google Scholar] [CrossRef] [PubMed]
- Tönshoff, C.; Bettinger, H.F. Pushing the limits of acene chemistry: The recent surge of large acenes. Chem. Eur. J. 2021, 27, 3193–3212. [Google Scholar] [CrossRef] [PubMed]
- Gupta, D.; Omont, A.; Bettinger, H.F. Energetics of formation of cyclacenes from 2,3-didehydroacenes and implications for astrochemistry. Chem. Eur. J. 2021, 27, 4605–4616. [Google Scholar] [CrossRef]
- Nieman, R.; Carvalho, J.R.; Jayee, B.; Hansen, A.; Aquino, A.J.; Kertesz, M.; Lischka, H. Polyradical character assessment using multireference calculations and comparison with density-functional derived fractional occupation number weighted density analysis. Phys. Chem. Chem. Phys. 2023, 25, 27380–27393. [Google Scholar] [CrossRef]
- Hanson-Heine, M.W.D. Static correlation in vibrational frequencies studied using thermally-assisted-occupation density functional theory. Chem. Phys. Lett. 2020, 739, 137012. [Google Scholar] [CrossRef]
- Hanson-Heine, M.W.D. Static electron correlation in anharmonic molecular vibrations: A hybrid TAO-DFT study. J. Phys. Chem. A 2022, 126, 7273–7282. [Google Scholar] [CrossRef]
- Su, Y.; Wang, X.; Wang, L.; Zhang, Z.; Wang, X.; Song, Y.; Power, P.P. Thermally controlling the singlet–triplet energy gap of a diradical in the solid state. Chem. Sci. 2016, 7, 6514–6518. [Google Scholar] [CrossRef]
- Yu, L.; Wu, Z.; Xie, G.; Zhong, C.; Zhu, Z.; Cong, H.; Ma, D.; Yang, C. Achieving a balance between small singlet–triplet energy splitting and high fluorescence radiative rate in a quinoxaline-based orange-red thermally activated delayed fluorescence emitter. Chem. Commun. 2016, 52, 11012–11015. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.B.; Michl, J. Singlet fission. Chem. Rev. 2010, 110, 6891–6936. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liu, Q.; Feng, W.; Sun, Y.; Li, F. Upconversion luminescent materials: Advances and applications. Chem. Rev. 2015, 115, 395–465. [Google Scholar] [CrossRef] [PubMed]
- Romero, N.A.; Nicewicz, D.A. Organic photoredox catalysis. Chem. Rev. 2016, 116, 10075–10166. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Sanders, S.N.; Cheng, W.; Low, J.Z.; Liu, J.; Campos, L.M.; Sun, T. Singlet fission: Progress and prospects in solar cells. Adv. Mater. 2017, 29, 1601652. [Google Scholar] [CrossRef] [PubMed]
- Rivero, P.; Jiménez-Hoyos, C.A.; Scuseria, G.E. Entanglement and polyradical nature of polycyclic aromatic hydrocarbons predicted by projected Hartree–Fock theory. J. Phys. Chem. B 2013, 117, 12750–12758. [Google Scholar] [CrossRef] [PubMed]
- Löwdin, P.-O.; Shull, H. Natural orbitals in the quantum theory of two-electron systems. Phys. Rev. 1956, 101, 1730–1739. [Google Scholar] [CrossRef]
- Takatsuka, K.; Fueno, T.; Yamaguchi, K. Distribution of odd electrons in ground state molecules. Theor. Chim. Acta 1978, 48, 175–183. [Google Scholar] [CrossRef]
- Head-Gordon, M. Characterizing unpaired electrons from the one-particle density matrix. Chem. Phys. Lett. 2003, 372, 508–511. [Google Scholar] [CrossRef]
- Shao, Y.; Gan, Z.; Epifanovsky, E.; Gilbert, A.T.; Wormit, M.; Kussmann, J.; Lange, A.W.; Behn, A.; Deng, J.; Feng, X.; et al. Advances in molecular quantum chemistry contained in the Q-Chem 4 program package. Mol. Phys. 2015, 113, 184–215. [Google Scholar] [CrossRef]















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seenithurai, S.; Chai, J.-D. Electronic Properties of Graphene Nano-Parallelograms: A Thermally Assisted Occupation DFT Computational Study. Molecules 2024, 29, 349. https://doi.org/10.3390/molecules29020349
Seenithurai S, Chai J-D. Electronic Properties of Graphene Nano-Parallelograms: A Thermally Assisted Occupation DFT Computational Study. Molecules. 2024; 29(2):349. https://doi.org/10.3390/molecules29020349
Chicago/Turabian StyleSeenithurai, Sonai, and Jeng-Da Chai. 2024. "Electronic Properties of Graphene Nano-Parallelograms: A Thermally Assisted Occupation DFT Computational Study" Molecules 29, no. 2: 349. https://doi.org/10.3390/molecules29020349
APA StyleSeenithurai, S., & Chai, J. -D. (2024). Electronic Properties of Graphene Nano-Parallelograms: A Thermally Assisted Occupation DFT Computational Study. Molecules, 29(2), 349. https://doi.org/10.3390/molecules29020349