
Synthesis and Dynamic Behavior of Ce(IV) Double-Decker Complexes of Sterically Hindered Phthalocyanines


Abstract
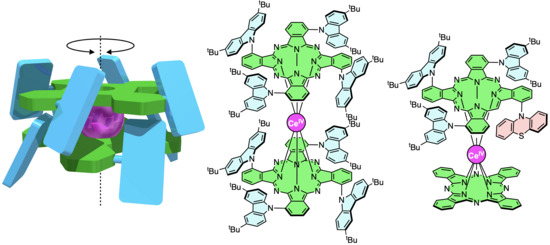
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Molecular Design of Sterically Hindered Double-Decker Complexes
2.2. Synthesis of the Pc Ligands Functionalized at the -Positions with Bulky Groups
2.2.1. Symmetric A4 Pc Ligands H2Pc1 and H2Pc2
2.2.2. Disymmetric A3B Pc Ligand H2Pc3
2.3. Synthesis of the Double-Decker Cerium(IV) Complexes
2.3.1. Homoleptic Double Deckers CeIV(Pc2)2 and CeIV(Pc3)2
2.3.2. Heteroleptic Double-Decker CeIV(Pc)(Pc3)
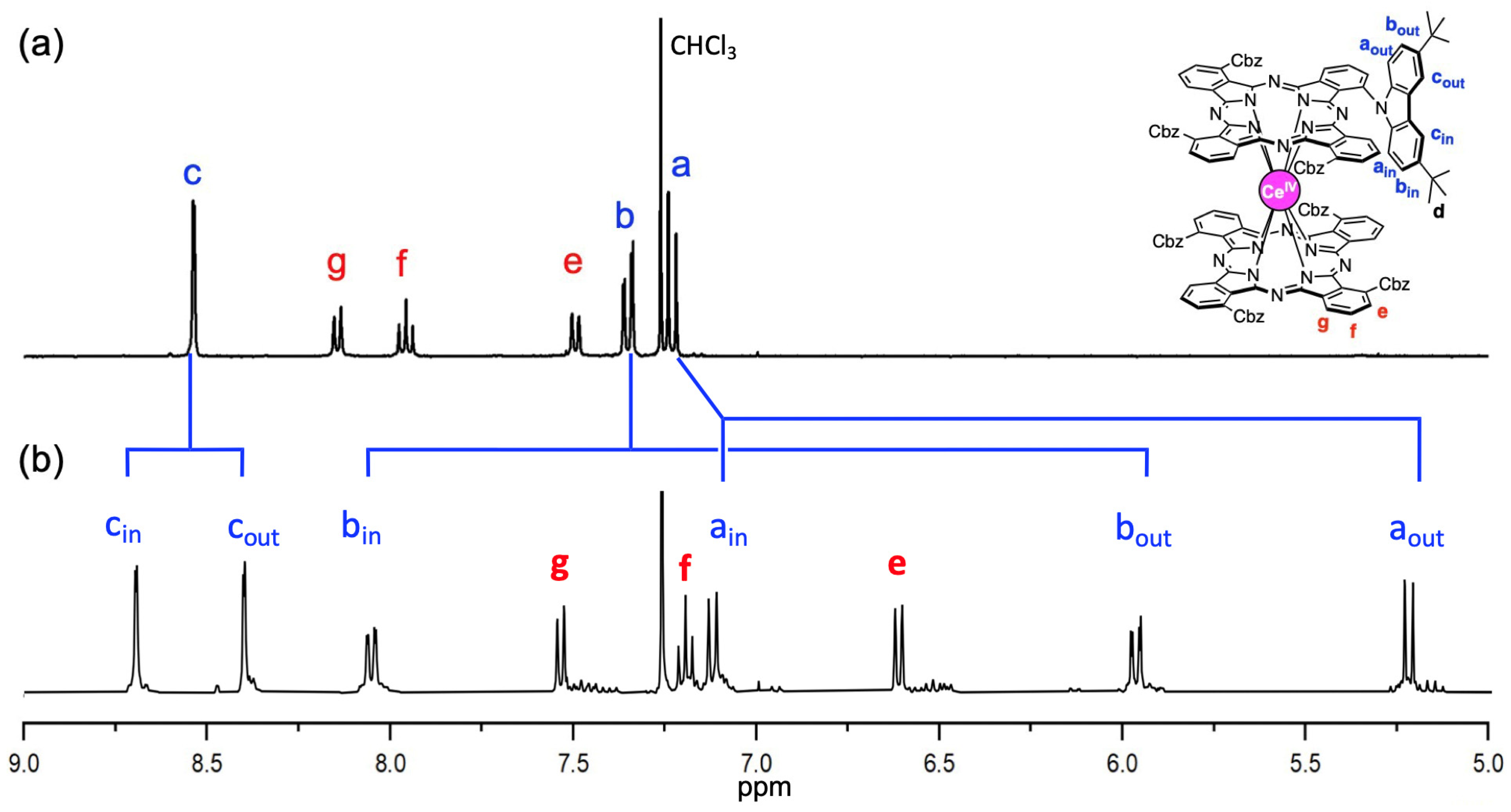
2.4. 1H-NMR Studies of the Dynamic Internal Rotating Motions in CeIV(Pc)(Pc3)
3. Materials and Methods
3.1. General Informations
3.2. Synthesis
3.2.1. Homoleptic Cerium(IV) Double-Decker Complex CeIV(Pc2)2
3.2.2. Homoleptic Cerium(IV) Double-Decker Complex CeIV(Pc3)2
3.2.3. Heteroleptic Cerium(IV) Double-Decker Complex CeIV(Pc)(Pc3)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Claessens, C.G.; Hahn, U.; Torres, T. Phthalocyanines: From outstanding electronic properties to emerging applications. Chem. Rec. 2008, 8, 75–97. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Calvete, M.J.F. Phthalocyanines: An old dog can still have new (photo)tricks! Molecules 2021, 26, 2823. [Google Scholar] [CrossRef]
- Kong, X.; Zhang, X.; Gao, D.; Qi, D.; Chen, Y.; Jiang, J. Air-stable ambipolar field-effect transistor based on a solution-processed octanaphthoxy- substituted tris(phthalocyaninato) europium semiconductor with high and balanced carrier mobilities. Chem. Sci. 2015, 6, 1967–1972. [Google Scholar] [CrossRef]
- Pekbelgin Karaoğlu, H.; Kalkan Burat, A. α- and β-substituted metal-free phthalocyanines: Synthesis, photophysical and electrochemical properties. Molecules 2020, 25, 363. [Google Scholar] [CrossRef] [PubMed]
- Bottari, G.; de la Torre, G.; Guldi, D.M.; Torres, T. Covalent and noncovalent phthalocyanine-carbon nanostructure systems: Synthesis, photoinduced electron transfer, and application to molecular photovoltaics. Chem. Rev. 2010, 110, 6768–6816. [Google Scholar] [CrossRef] [PubMed]
- Sorokin, A.B. Phthalocyanine Metal Complexes in Catalysis. Chem. Rev. 2013, 113, 8152–8191. [Google Scholar] [CrossRef] [PubMed]
- Martynov, A.G.; Horii, Y.; Katoh, K.; Bian, Y.; Jiang, J.; Yamashita, M.; Gorbunova, Y.G. Rare-earth based tetrapyrrolic sandwiches: Chemistry, materials and applications. Chem. Soc. Rev. 2022, 51, 9262–9339. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Ng, D.K.P. A decade journey in the chemistry of sandwich-type tetrapyrrolato—Rare earth complexes. Acc. Chem. Res. 2009, 42, 79–88. [Google Scholar] [CrossRef] [PubMed]
- Chabach, D.; Tahiri, M.; Cian, A.D.; Fischer, J.; Weiss, R.; El Malouli Bibout, E. Tervalent-Metal Porphyrin-Phthalocyanine Heteroleptic Sandwich-Type Complexes. Synthesis, Structure, and Spectroscopic Characterization of Their Neutral, Singly-Oxidized, and Singly-Reduced States. J. Am. Chem. Soc. 1995, 117, 8548–8556. [Google Scholar] [CrossRef]
- Jiang, J.; Bian, Y.; Furuya, F.; Liu, W.; Choi, M.T.M.; Kobayashi, N.; Li, H.-W.; Yang, Q.; Mak, T.C.W.; Ng, D.K.P. Synthesis, Structure, Spectroscopic Properties, and Electrochemistry of Rare Earth Sandwich Compounds with Mixed 2,3-Naphthalocyaninato and Octaethylporphyrinato Ligands. Chem. Eur. J. 2001, 7, 5059–5069. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Jiang, J.; Tao, Y.; Choi, M.T.M.; Li, R.; Ng, A.C.H.; Zhu, P.; Pan, N.; Sun, X.; Arnold, D.P.; et al. Tuning the Valence of the Cerium Center in (Na)phthalocyaninato and Porphyrinato Cerium Double-Deckers by Changing the Nature of the Tetrapyrrole Ligands. J. Am. Chem. Soc. 2003, 125, 12257–12267. [Google Scholar] [CrossRef]
- Stepanow, S.; Honolka, J.; Gambardella, P.; Vitali, L.; Abdurakhmanova, N.; Tseng, T.; Rauschenbach, S.; Tait, S.L.; Sessi, V.; Klyatskaya, S.; et al. Spin and orbital magnetic moment anisotropies of monodispersed bis(phthalocyaninato)terbium on a copper surface. J. Am. Chem. Soc. 2010, 132, 11900–11901. [Google Scholar] [CrossRef] [PubMed]
- Mele, G.; Garcìa-Lòpez, E.; Palmisano, L.; Dyrda, G.; Słota, R. Photocatalytic Degradation of 4-Nitrophenol in Aqueous Suspension by Using Polycrystalline TiO2 Impregnated with Lanthanide Double-Decker Phthalocyanine Complexes. J. Phys. Chem. C 2007, 111, 6581–6588. [Google Scholar] [CrossRef]
- Gisbert, Y.; Abid, S.; Kammerer, C.; Rapenne, G. Molecular gears from solution to surfaces. Chem. Eur. J. 2021, 27, 12019–12031. [Google Scholar] [CrossRef]
- Zhang, Y.; Kersell, H.; Stefak, R.; Echeverria, J.; Iancu, V.; Perera, G.; Li, Y.; Braun, K.F.; Joachim, C.; Rapenne, G.; et al. Simultaneous and coordinated rotational switching of all molecular rotors in a network. Nat. Nanotech. 2016, 11, 706–712. [Google Scholar] [CrossRef]
- Michl, J.; Sykes, E.C.H. Molecular Rotors and Motors: Recent Advances and Future Challenges. ACS Nano 2009, 3, 1042–1048. [Google Scholar] [CrossRef]
- Ikeda, M.; Takeuchi, M.; Shinkai, S.; Tani, F.; Naruta, Y. Synthesis of new diaryl-substituted triple-decker and tetraaryl-substituted double-decker lanthanum(III) porphyrins and their porphyrin ring rotational speed as compared with that of double-decker cerium(IV) porphyrins. Bull. Chem. Soc. Jpn. 2001, 74, 739–746. [Google Scholar] [CrossRef]
- Miyake, K.; Fukuta, M.; Asakawa, M.; Hori, Y.; Ikeda, T.; Shimizu, T. Molecular motion of surface-immobilized double-decker phthalocyanine complexes. J. Am. Chem. Soc. 2009, 131, 17808–17813. [Google Scholar] [CrossRef]
- Ecija, D.; Auwärter, W.; Vijayaraghavan, S.; Seufert, K.; Bischoff, F.; Tashiro, K.; Barth, J.V. Assembly and manipulation of rotatable cerium porphyrinato sandwich complexes on a surface. Angew. Chem. Int. Ed. 2011, 50, 3872–3877. [Google Scholar] [CrossRef]
- Otsuki, J.; Taka, M.; Kobayashi, D. Rotational libration of a porphyrin/phthalocyanine double-decker complex with Ce(IV) as revealed by 1H NMR and STM. Chem. Lett. 2011, 40, 717–719. [Google Scholar] [CrossRef]
- Martynov, A.G.; Kirakosyan, G.A. Characterizing the conformational behavior of yttrium(III) tris-phthalocyaninates using VT-NMR spectroscopy. Macroheterocycles 2023, 16, 150–155. [Google Scholar] [CrossRef]
- Yamamoto, S.; Kuribayashi, K.; Murakami, T.N.; Kwon, E.; Stillman, M.J.; Kobayashi, N.; Segawa, H.; Kimura, M. Regioregular phthalocyanines substituted with bulky donors at non-peripheral positions. Chem. Eur. J. 2017, 23, 15446–15454. [Google Scholar] [CrossRef]
- Wang, R.; Li, Y.; Li, R.; Cheng, D.Y.Y.; Zhu, P.; Ng, D.K.P.; Bao, M.; Cui, X.; Kobayashi, N.; Jiang, J. Heteroleptic rare earth double-decker complexes with naphthalocyaninato and phthalocyaninato ligands. General synthesis, spectroscopic, and electrochemical characteristics. Inorg. Chem. 2005, 44, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, R.; Li, Y.; Zhang, X.; Zhu, P.; Lo, P.C.; Ng, D.K.P.; Pan, N.; Ma, C.; Kobayashi, N.; et al. Controlling the nature of mixed (phthalocyaninato)(porphyrinato) rare-earth(III) double-decker complexes: The Effects of nonperipheral alkoxy substitution of the phthalocyanine ligand. Chem. Eur. J. 2006, 12, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Also for heteroleptic complexes Martynov, A.G.; Polovkova, M.A.; Berezhnoy, G.S.; Sinelshchikova, A.A.; Khrustalev, V.N.; Birin, K.P.; Kirakosyan, G.A.; Gorbunova, Y.G.; Tsivadze, A.Y. Heteroleptic Crown-Substituted Tris(phthalocyaninates) as Dynamic Supramolecular Scaffolds with Switchable Rotational States and Tunable Magnetic Properties. Inorg. Chem. 2021, 60, 9110–9121. [Google Scholar] [CrossRef] [PubMed]
- Birin, K.P.; Gorbunova, Y.G.; Tsivadze, A.Y. NMR investigation of intramolecular dynamics of heteroleptic triple-decker (porphyrinato)(phthalocyaninato) lanthanides. Dalton Trans. 2011, 40, 11474–11479. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zhang, X.; Zhu, P.; Ng, D.K.P.; Kobayashi, N.; Jiang, J. Electron-donating or -withdrawing nature of substituents revealed by the electrochemistry of metal-free phthalocyanines. Inorg. Chem. 2006, 45, 2327–2334. [Google Scholar] [CrossRef]
- Sych, G.; Pashazadeh, R.; Danyliv, Y.; Bezvikonnyi, O.; Volyniuk, D.; Lazauskas, A.; Grazulevicius, J.V. Reversibly switchable phase-dependent emission of quinoline and phenothiazine derivatives towards applications in optical sensing and information multicoding. Chem. Eur. J. 2021, 27, 2826–2836. [Google Scholar] [CrossRef]
- Schoch, T.D.; Mondal, M.; Weaver, J.D. Catalyst-free hydrodefluorination of perfluoroarenes with NaBH4. Org. Lett. 2021, 23, 1588–1593. [Google Scholar] [CrossRef]
- Pushkarev, V.E.; Tolbin, A.Y.; Borisova, N.E.; Trashin, S.A.; Tomilova, L.G. A3B-type phthalocyanine-based homoleptic lanthanide(III) double-decker π-radical complexes bearing functional hydroxy groups: Synthetic approach, spectral properties and electrochemical study. Eur. J. Inorg. Chem. 2010, 5254–5262. [Google Scholar] [CrossRef]
- Göktuğ, Ö.; Soganci, T.; Ak, M.; Şener, M.K. Efficient synthesis of EDOT modified ABBB-type unsymmetrical zinc phthalocyanine: Optoelectrochromic and glucose sensing properties of its copolymerized film. New J. Chem. 2017, 41, 14080–14087. [Google Scholar] [CrossRef]
- Jin, H.-G.; Jiang, X.; Kühne, I.A.; Clair, S.; Monnier, V.; Chendo, C.; Novitchi, G.; Powell, A.K.; Kadish, K.M.; Balaban, T.S. Microwave-mediated synthesis of bulky lanthanide porphyrin–phthalocyanine triple-deckers: Electrochemical and magnetic properties. Inorg. Chem. 2017, 56, 4864–4873. [Google Scholar] [CrossRef] [PubMed]
- Yamada, Y.; Nakajima, H.; Kobayashi, C.; Shuku, Y.; Awaga, K.; Akine, S.; Tanaka, K. Synthesis of isomeric Tb3+-phthalocyanine double-decker complexes depending on the difference in the direction of coordination plane and their magnetic properties. Chem. Eur. J. 2023, 29, e202203272. [Google Scholar] [CrossRef]
- Zhang, X.; Cai, X.; Qi, D.; Yao, P.; Bian, Y.; Jiang, J. Methyloxy substituted heteroleptic bis(phthalocyaninato) yttrium complexes: Density functional calculations. ChemPhysChem 2008, 9, 781–792. [Google Scholar] [CrossRef] [PubMed]
- Davoras, E.M.; Spyroulias, G.A.; Mikros, E.; Coutsolelos, A.G. Intramolecular dynamics of asymmetric lanthanide(III) porphyrin sandwich complexes in solution. 1H-NMR spectroscopic elucidation of stereochemical effects of substituted cerium porphyrin double-deckers. Inorg. Chem. 1994, 33, 3430–3434. [Google Scholar] [CrossRef]
- Babailov, S.P.; Coutsolelos, A.G.; Dikiy, A.; Spyroulias, G.A. Intramolecular Dynamics of Asymmetric Lanthanide(III) Porphyrin Sandwich Complexes in Solution. Eur. J. Inorg. Chem. 2001, 2001, 303–306. [Google Scholar] [CrossRef]
- Pernin, D.; Haberroth, K.; Simon, J. Novel unsymmetrical monofunctionalized lutetium and dysprosium bisphthalocyanines with seven crown-ether units and one hexyl hexanoate side-group. J. Chem. Soc. Perkin 1 1997, 1265–1266. [Google Scholar] [CrossRef]
- Sheng, N.; Li, R.; Choi, C.-F.; Su, W.; Ng, D.K.P.; Cui, X.; Yoshida, K.; Kobayashi, N.; Jiang, J. Heteroleptic bis(phthalocyaninato) europium(III) complexes fused with different numbers of 15-crown-5 moieties. Synthesis, spectroscopy, electrochemistry, and supramolecular structure. Inorg. Chem. 2006, 45, 3794–3802. [Google Scholar] [CrossRef]
- Ballesteros, B.; de la Torre, G.; Shearer, A.; Hausmann, A.; Herranz, M.Á.; Guldi, D.M.; Torres, T. Lanthanide(III) bis(phthalocyaninato)–[60]fullerene dyads: Synthesis, characterization, and photophysical properties. Chem. Eur. J. 2010, 16, 114–125. [Google Scholar] [CrossRef]
- Tashiro, K.; Konishi, K.; Aida, T. Metal bisporphyrinate double-decker complexes as redox-responsive rotating modules. Studies on ligand rotation activities of the reduced and oxidized forms using chirality as a Probe. J. Am. Chem. Soc. 2000, 122, 7921–7926. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subramaniam, J.D.; Nishino, T.; Yasuhara, K.; Rapenne, G. Synthesis and Dynamic Behavior of Ce(IV) Double-Decker Complexes of Sterically Hindered Phthalocyanines. Molecules 2024, 29, 888. https://doi.org/10.3390/molecules29040888
Subramaniam JD, Nishino T, Yasuhara K, Rapenne G. Synthesis and Dynamic Behavior of Ce(IV) Double-Decker Complexes of Sterically Hindered Phthalocyanines. Molecules. 2024; 29(4):888. https://doi.org/10.3390/molecules29040888
Chicago/Turabian StyleSubramaniam, Jeevithra Dewi, Toshio Nishino, Kazuma Yasuhara, and Gwénaël Rapenne. 2024. "Synthesis and Dynamic Behavior of Ce(IV) Double-Decker Complexes of Sterically Hindered Phthalocyanines" Molecules 29, no. 4: 888. https://doi.org/10.3390/molecules29040888
APA StyleSubramaniam, J. D., Nishino, T., Yasuhara, K., & Rapenne, G. (2024). Synthesis and Dynamic Behavior of Ce(IV) Double-Decker Complexes of Sterically Hindered Phthalocyanines. Molecules, 29(4), 888. https://doi.org/10.3390/molecules29040888