

Identification of Phytochemicals from Arabian Peninsula Medicinal Plants as Strong Binders to SARS-CoV-2 Proteases (3CLPro and PLPro) by Molecular Docking and Dynamic Simulation Studies

 ,
, 

Abstract
:1. Introduction
2. Results
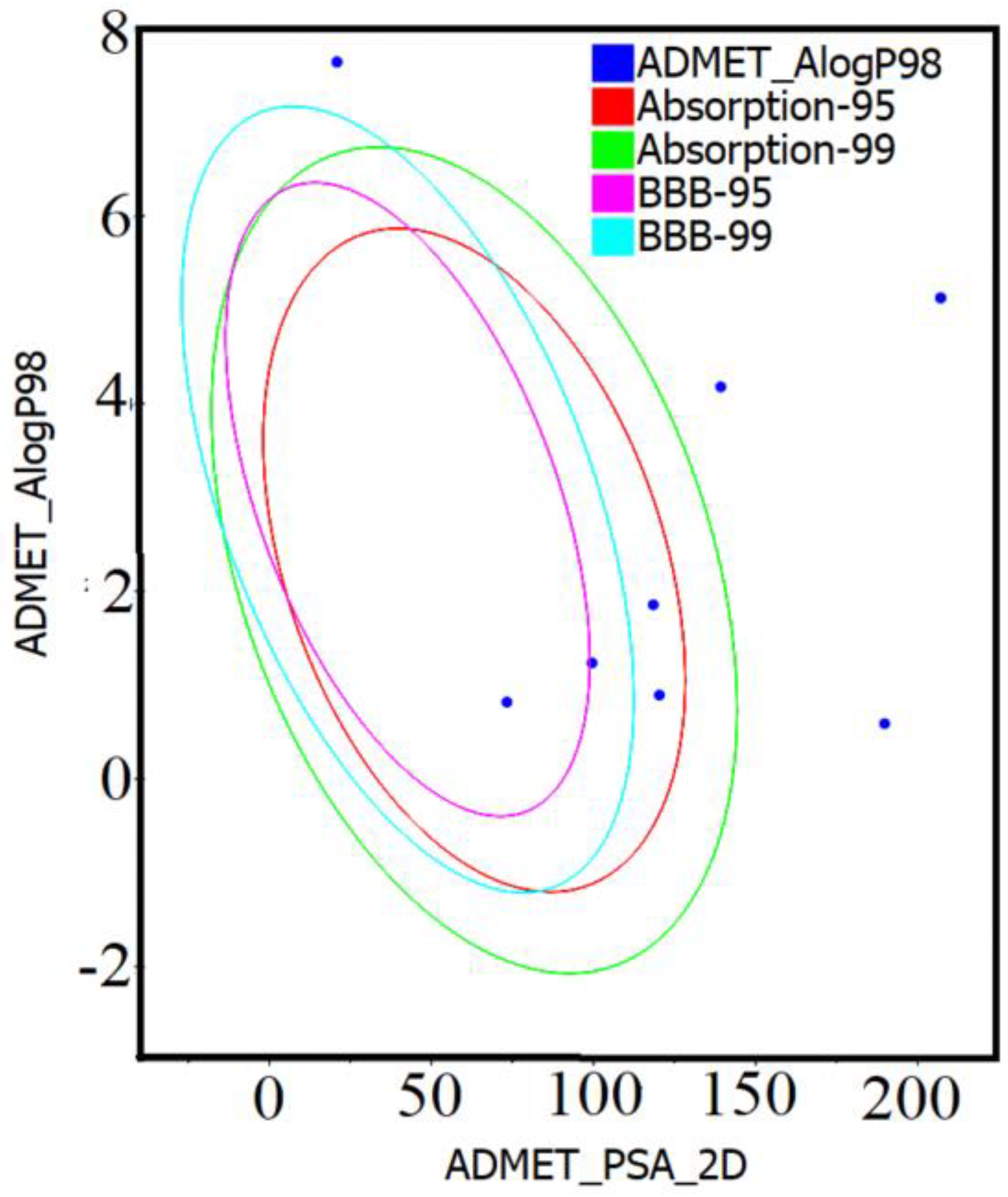
2.1. Drug-Likeness and ADMET Analysis of Compounds 1–10
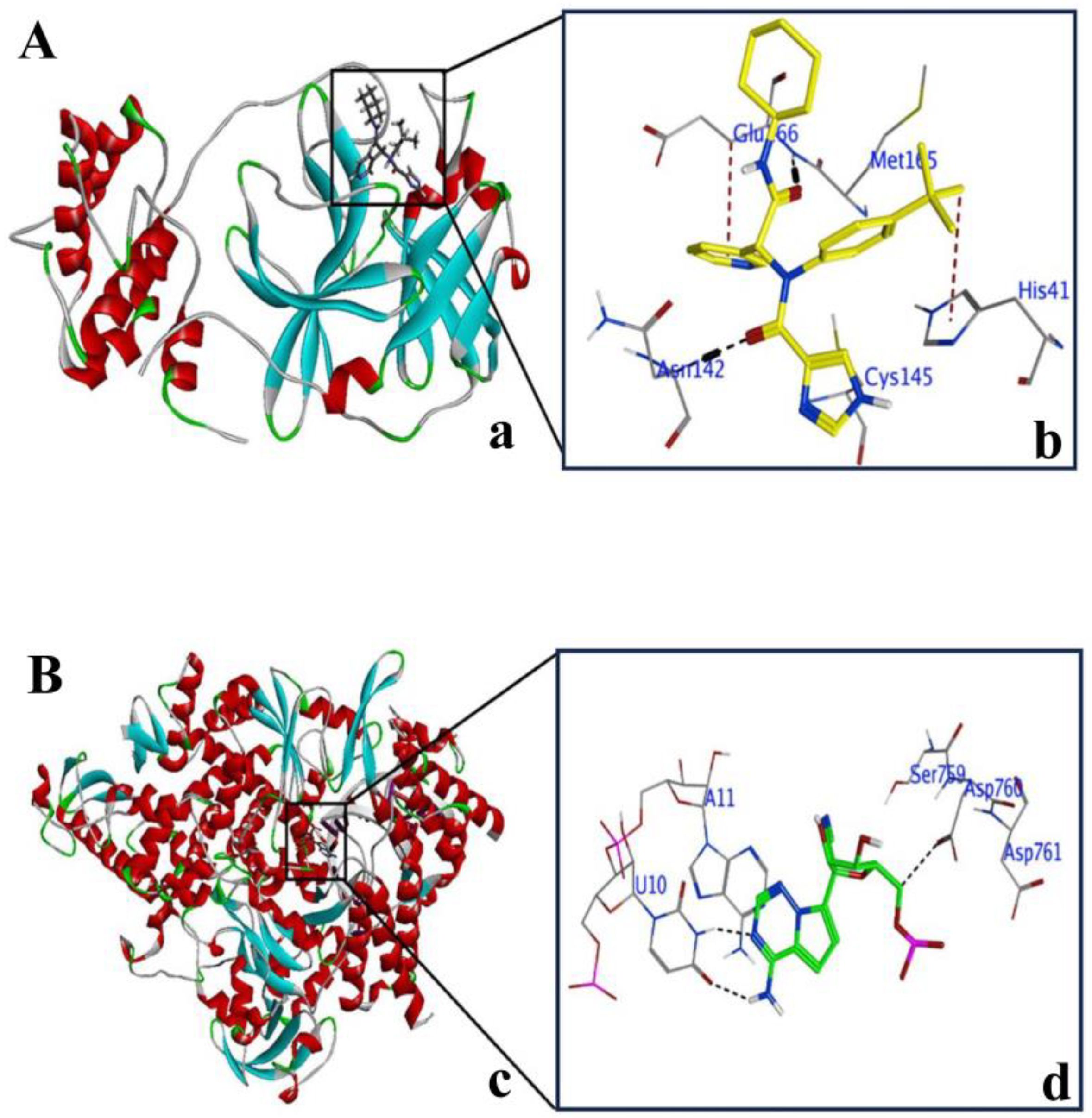
2.2. Explanation of Active Sites in 3CLPro and PLPro
2.3. Validation of Docking Protocol with 3CLPro and PLPro
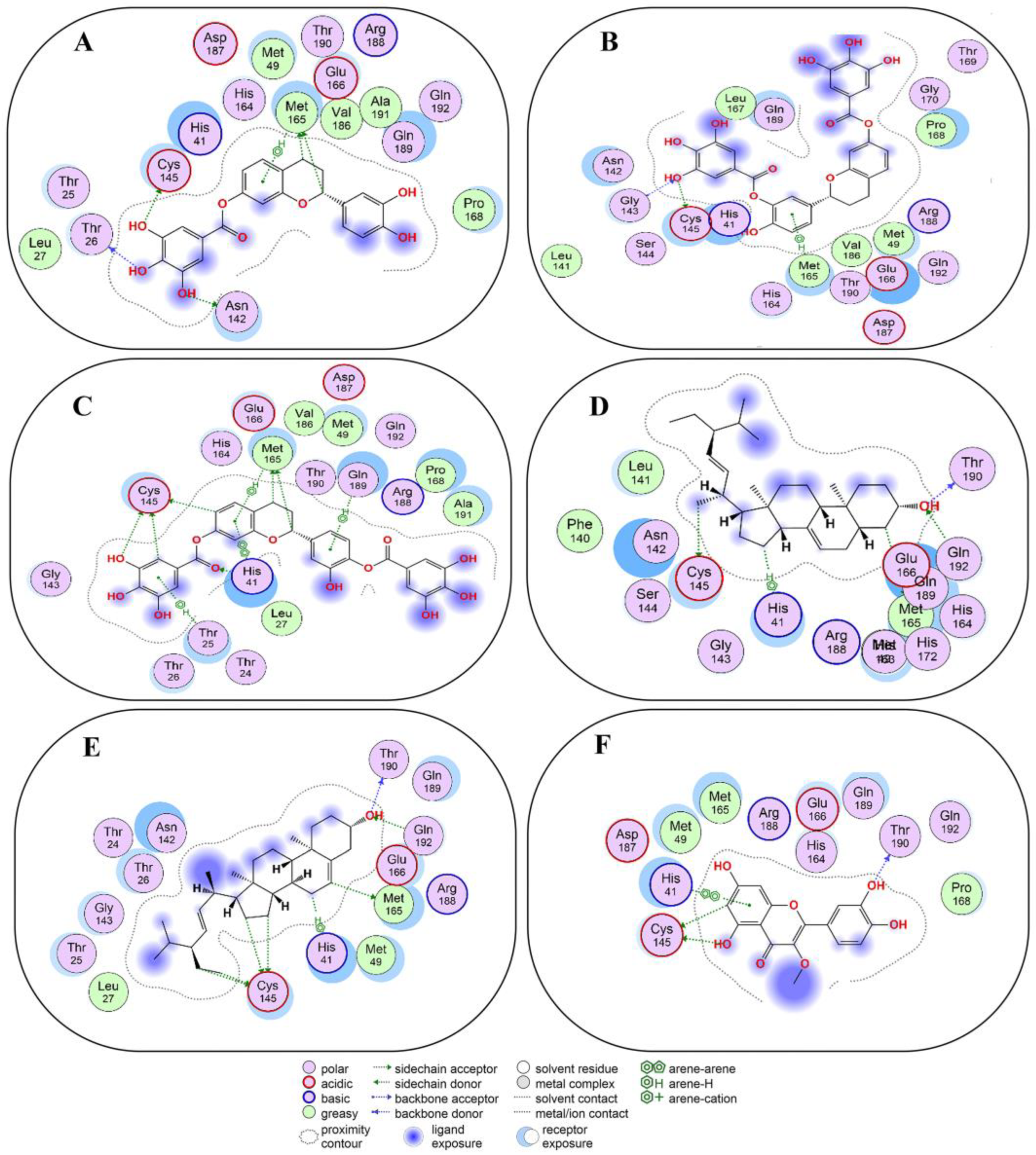
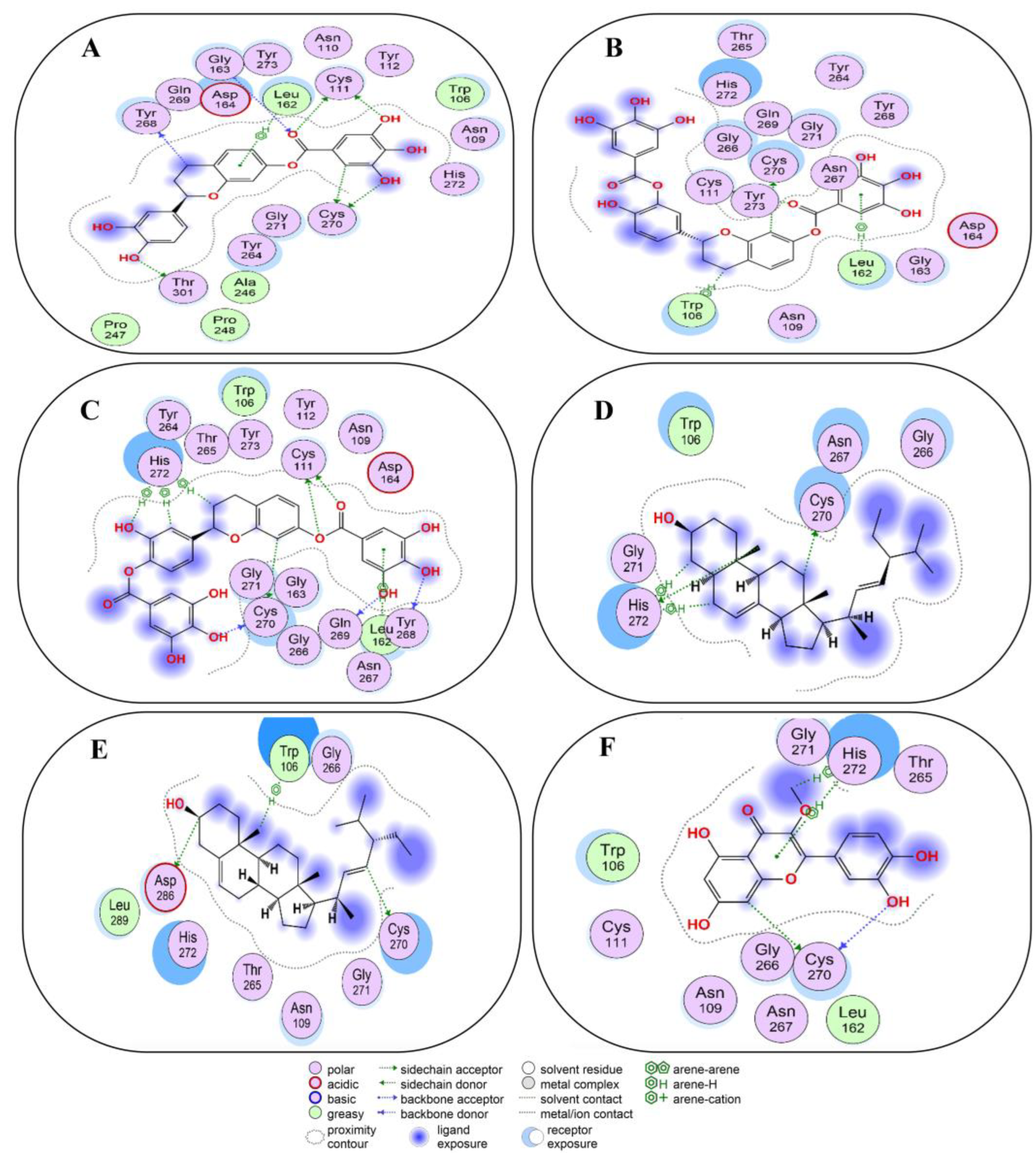
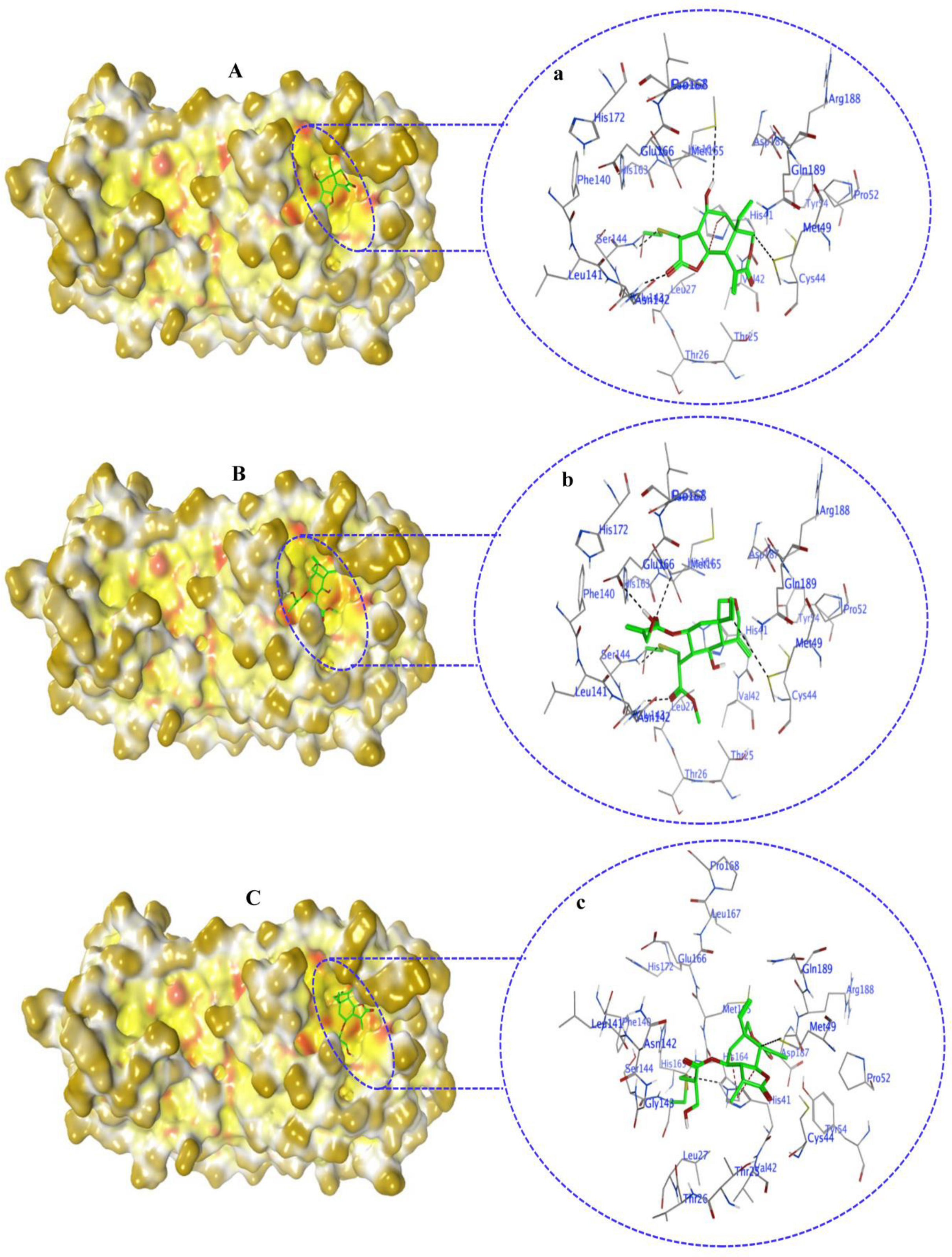
2.4. Non-Covalent Docking of Compounds 1–10 with 3CLPro
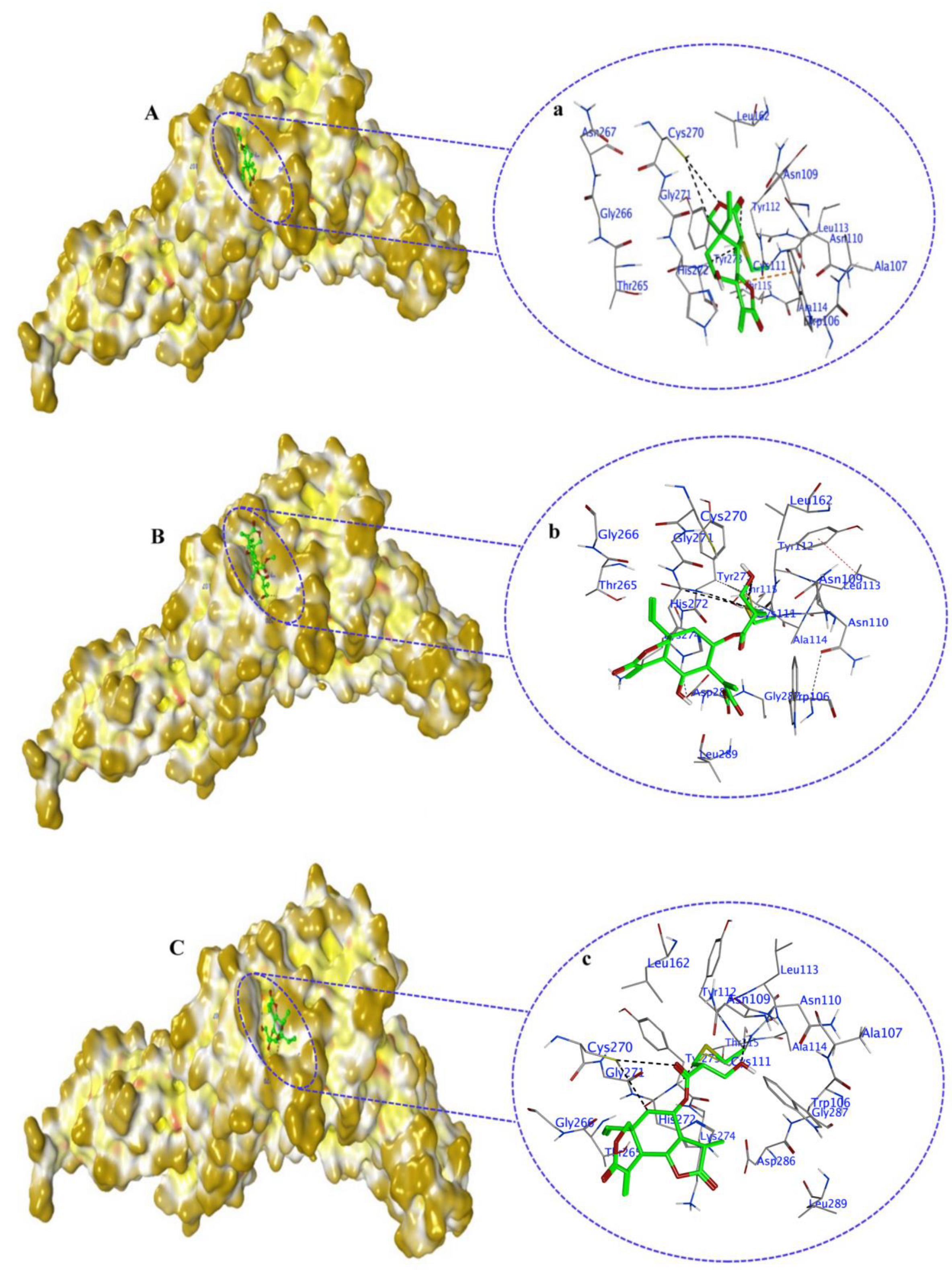
2.5. Non-Covalent Docking of Compounds 1–10 PLPro
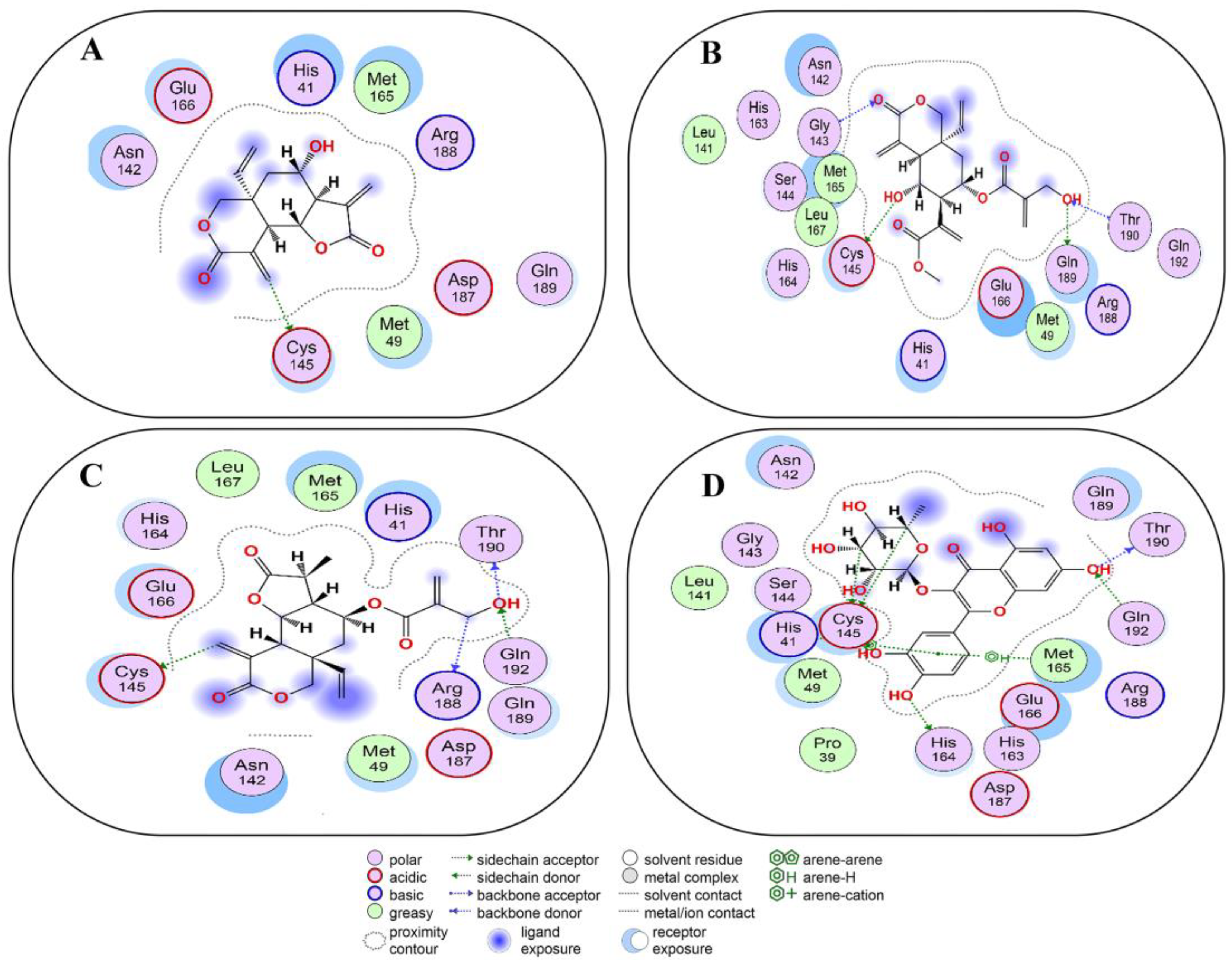
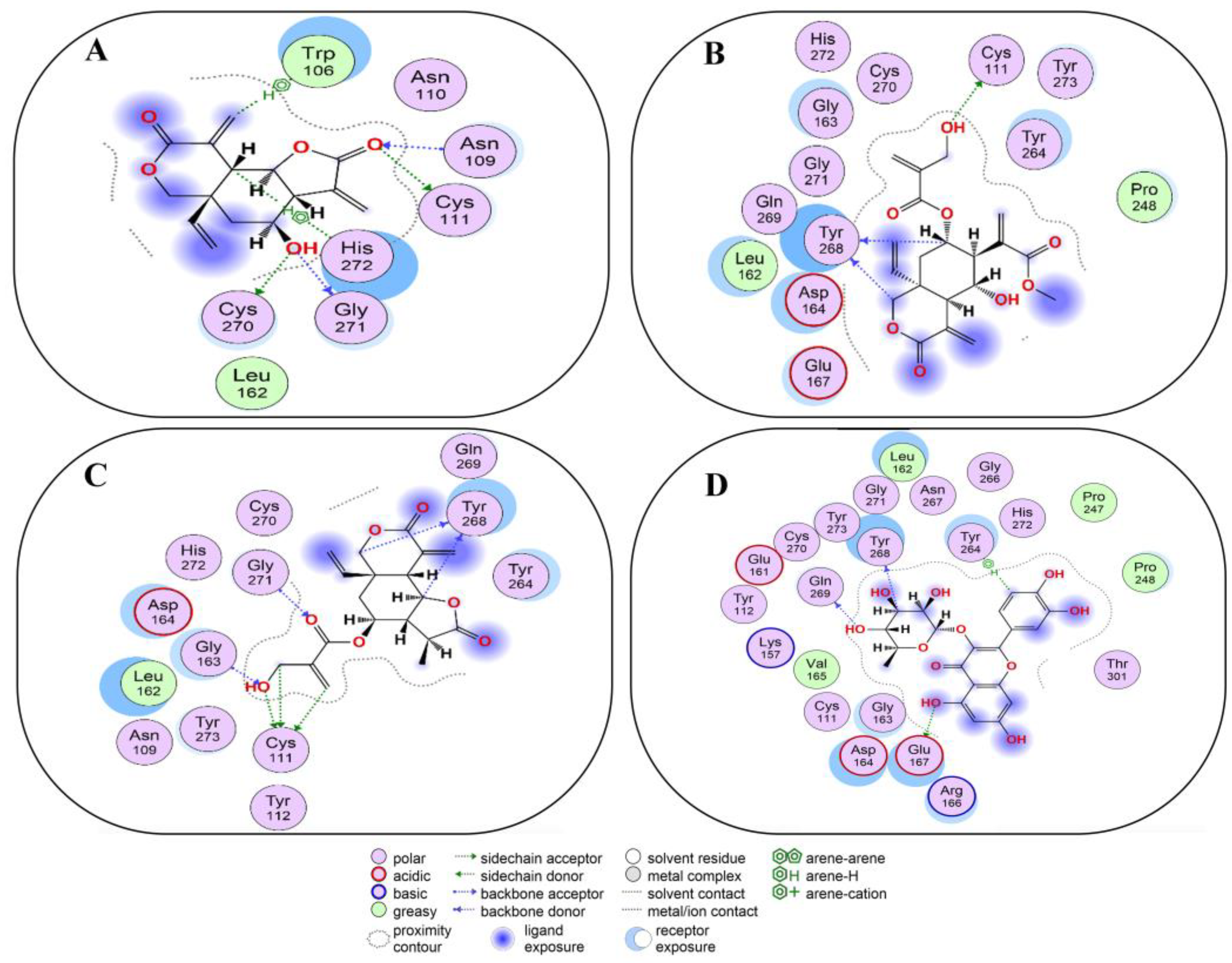
2.6. Covalent Docking of Compounds 1–10 3CLPro
2.7. Covalent Docking of Compounds 1–10 with PLPro
2.8. MD Simulation Analysis
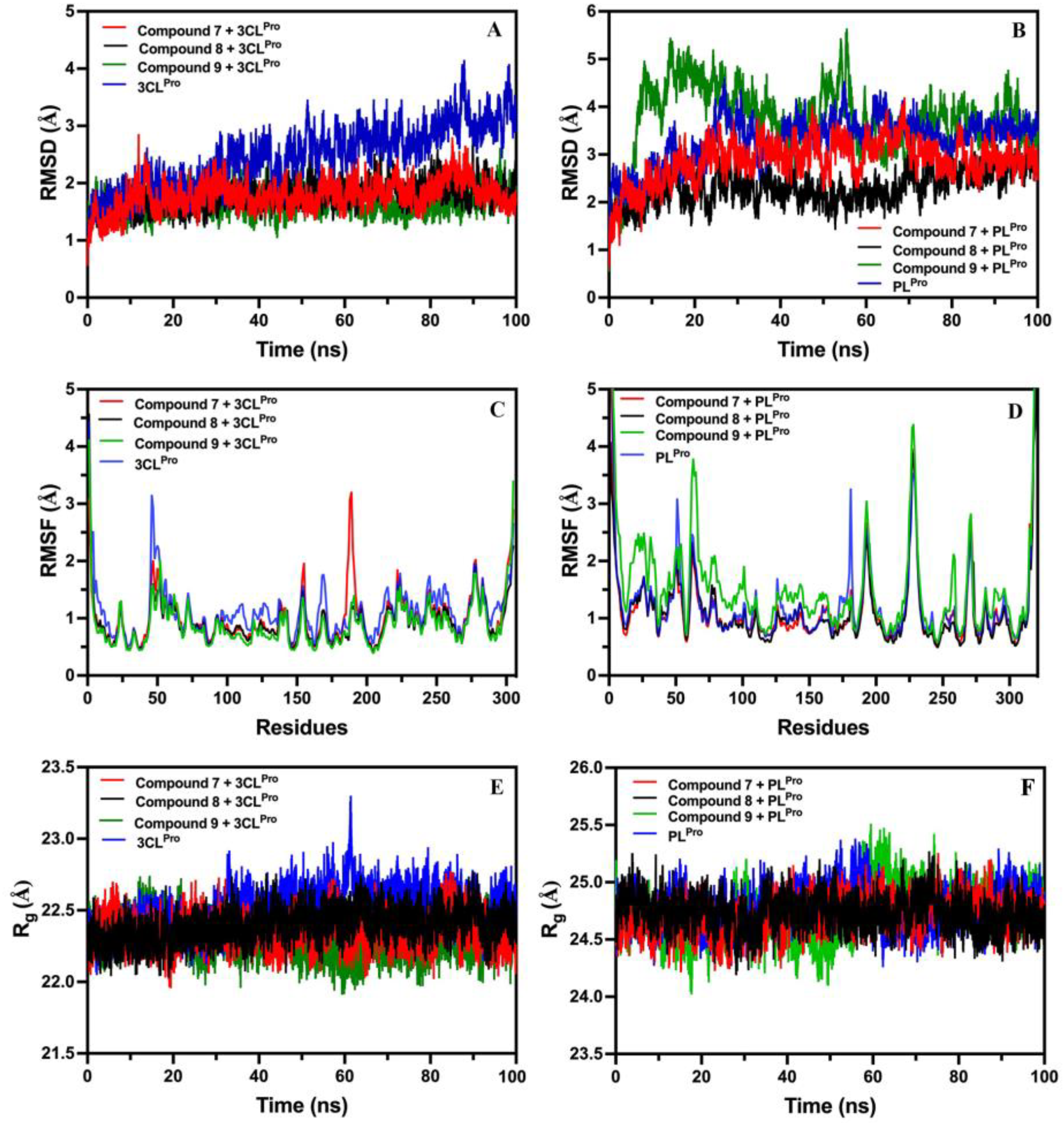
2.8.1. Root-Mean-Square-Deviation (RMSD) of Compounds 7, 8, and 9 with 3CLPro and PLPro
2.8.2. Root-Mean-Square-Fluctuation (RMSF) of Compounds 7, 8, and 9 with 3CLPro and PLPro
2.8.3. Radius of Gyration (Rg) of Compounds 7, 8, and 9 with 3CLPro and PLPro
3. Discussion
4. Materials and Methods
4.1. Phytochemicals from Medicinal Plants
4.2. Drug-Likeness and ADMET Properties
4.3. Ligand Preparation
4.4. Target Protein Preparation
4.5. Docking Experiments
4.5.1. Validation of Docking
4.5.2. Non-Covalent Docking
4.5.3. Covalent Docking
4.6. Molecular Dynamics (MD) Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. WHO Coronavirus (COVID-19) Dashboard. 2023. Available online: https://covid19.who.int (accessed on 22 November 2023).
- Guan, W.-J.; Ni, Z.-Y.; Hu, Y.; Liang, W.-H.; Ou, C.-Q.; He, J.-X.; Liu, L.; Shan, H.; Lei, C.-L.; Hui, D.S.C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Bi, Q.; Wu, Y.; Mei, S.; Ye, C.; Zou, X.; Zhang, Z.; Liu, X.; Wei, L.; Truelove, S.A.; Zhang, T.; et al. Epidemiology and transmission of COVID-19 in 391 cases and 1286 of their close contacts in Shenzhen, China: A retrospective cohort study. Lancet Infect. Dis. 2020, 20, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Monteleone, G.; Sarzi-Puttini, P.C.; Ardizzone, S. Preventing COVID-19-induced pneumonia with anticytokine therapy. Lancet Rheumatol. 2020, 2, e255–e256. [Google Scholar] [CrossRef] [PubMed]
- Amaral-Machado, L.; Oliveira, W.N.; Rodrigues, V.M.; Albuquerque, N.A.; Alencar, É.N.; Egito, E.S.T. Could natural products modulate early inflammatory responses, preventing acute respiratory distress syndrome in COVID-19-confirmed patients? Biomed. Pharmacother. 2021, 134, 111143. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Geng, M.; Peng, Y.; Meng, L.; Lu, S. Molecular immune pathogenesis and diagnosis of COVID-19. J. Pharm. Anal. 2020, 10, 102–108. [Google Scholar] [CrossRef] [PubMed]
- Das, G.; Das, T.; Chowdhury, N.; Chatterjee, D.; Bagchi, A.; Ghosh, Z. Repurposed drugs and nutraceuticals targeting envelope protein: A possible therapeutic strategy against COVID-19. Genomics 2021, 113, 1129–1140. [Google Scholar] [CrossRef]
- Nile, S.H.; Nile, A.; Qiu, J.; Li, L.; Jia, X.; Kai, G. COVID-19: Pathogenesis, cytokine storm and therapeutic potential of interferons. Cytokine Growth Factor Rev. 2020, 53, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Perlman, S. Coronaviruses: An Overview of Their Replication and Pathogenesis. In Coronaviruses: Methods and Protocols; Maier, H.J., Bickerton, E., Britton, P., Eds.; Springer New York: New York, NY, USA, 2015; pp. 1–23. [Google Scholar] [CrossRef]
- Jade, D.; Ayyamperumal, S.; Tallapaneni, V.; Joghee Nanjan, C.M.; Barge, S.; Mohan, S.; Nanjan, M.J. Virtual high throughput screening: Potential inhibitors for SARS-CoV-2 PLPRO and 3CLPRO proteases. Eur. J. Pharmacol. 2021, 901, 174082. [Google Scholar] [CrossRef] [PubMed]
- Tumskiy, R.S.; Tumskaia, A.V.; Klochkova, I.N.; Richardson, R.J. SARS-CoV-2 proteases Mpro and PLpro: Design of inhibitors with predicted high potency and low mammalian toxicity using artificial neural networks, ligand-protein docking, molecular dynamics simulations, and ADMET calculations. Comput. Biol. Med. 2023, 153, 106449. [Google Scholar] [CrossRef]
- Nesterenko, P.A.; McLaughlin, J.; Tsai, B.L.; Burton Sojo, G.; Cheng, D.; Zhao, D.; Mao, Z.; Bangayan, N.J.; Obusan, M.B.; Su, Y.; et al. HLA-A∗02:01 restricted T cell receptors against the highly conserved SARS-CoV-2 polymerase cross-react with human coronaviruses. Cell Rep. 2021, 37, 110167. [Google Scholar] [CrossRef]
- Zhang, L.-c.; Zhao, H.-l.; Liu, J.; He, L.; Yu, R.-l.; Kang, C.-m. Design of SARS-CoV-2 Mpro, PLpro dual-target inhibitors based on deep reinforcement learning and virtual screening. Future Med. Chem. 2022, 14, 393–405. [Google Scholar] [CrossRef]
- Lin, C.-W.; Tsai, F.-J.; Tsai, C.-H.; Lai, C.-C.; Wan, L.; Ho, T.-Y.; Hsieh, C.-C.; Chao, P.-D.L. Anti-SARS coronavirus 3C-like protease effects of Isatis indigotica root and plant-derived phenolic compounds. Antivir. Res. 2005, 68, 36–42. [Google Scholar] [CrossRef]
- Prasansuklab, A.; Theerasri, A.; Rangsinth, P.; Sillapachaiyaporn, C.; Chuchawankul, S.; Tencomnao, T. Anti-COVID-19 drug candidates: A review on potential biological activities of natural products in the management of new coronavirus infection. J. Tradit. Complement. Med. 2020, 11, 144–157. [Google Scholar] [CrossRef]
- Morse, J.S.; Lalonde, T.; Xu, S.; Liu, W.R. Learning from the Past: Possible Urgent Prevention and Treatment Options for Severe Acute Respiratory Infections Caused by 2019-nCoV. ChemBioChem 2020, 21, 730–738. [Google Scholar] [CrossRef]
- Deng, W.; Xu, Y.; Kong, Q.; Xue, J.; Yu, P.; Liu, J.; Lv, Q.; Li, F.; Wei, Q.; Bao, L. Therapeutic efficacy of Pudilan Xiaoyan Oral Liquid (PDL) for COVID-19 in vitro and in vivo. Signal Transduct. Target. Ther. 2020, 5, 66. [Google Scholar] [CrossRef]
- Ni, L.; Zhou, L.; Zhou, M.; Zhao, J.; Wang, D.W. Combination of western medicine and Chinese traditional patent medicine in treating a family case of COVID-19. Front. Med. 2020, 14, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Schettig, R.; Sears, T.; Klein, M.; Tan-Lim, R.; Matthias, R., Jr.; Aussems, C.; Hummel, M.; Sears, R.; Poteet, Z.; Warren, D. COVID-19 Patient with Multifocal Pneumonia and Respiratory Difficulty Resolved Quickly: Possible Antiviral and Anti-Inflammatory Benefits of Quercinex (Nebulized Quercetin-NAC) as Adjuvant. Adv. Infect. Dis. 2020, 10, 45–55. [Google Scholar] [CrossRef]
- Dai, Y.; Qiang, W.; Gui, Y.; Tan, X.; Pei, T.; Lin, K.; Cai, S.; Sun, L.; Ning, G.; Wang, J.; et al. A large-scale transcriptional study reveals inhibition of COVID-19 related cytokine storm by traditional chinese medicines. Sci. Bull. 2021, 66, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.C.; Huang, Y.C.; Liaw, C.C.; Tsai, C.I.; Chiou, C.T.; Lin, C.J.; Wei, W.C.; Lin, S.J.; Tseng, Y.H.; Yeh, K.M.; et al. A traditional Chinese medicine formula NRICM101 to target COVID-19 through multiple pathways: A bedside-to-bench study. Biomed. Pharmacother. 2021, 133, 111037. [Google Scholar] [CrossRef]
- Shang, Z.P.; Yi, Y.; Yu, R.; Fan, J.J.; Huang, Y.X.; Qiao, X.; Ye, M. [Bioactive compounds of Jingfang Granules against SARS-CoV-2 virus proteases 3CL(pro) and PL(pro)]. Beijing Da Xue Xue Bao Yi Xue Ban 2022, 54, 907–919. [Google Scholar] [CrossRef] [PubMed]
- Guijarro-Real, C.; Plazas, M.; Rodríguez-Burruezo, A.; Prohens, J.; Fita, A. Potential In Vitro Inhibition of Selected Plant Extracts against SARS-CoV-2 Chymotripsin-Like Protease (3CLPro) Activity. Foods 2021, 10, 1503. [Google Scholar] [CrossRef]
- Ávila-Gálvez, M.Á.; Rafael-Pita, C.; Fernández, N.; Baixinho, J.; Anastácio, J.D.; Cankar, K.; Bosch, D.; Nunes dos Santos, C. Targeting proteases involved in the viral replication of SARS-CoV-2 by sesquiterpene lactones from chicory (Cichorium intybus L.). Food Funct. 2022, 13, 8977–8988. [Google Scholar] [CrossRef]
- Marín-Palma, D.; Tabares-Guevara, J.H.; Zapata-Cardona, M.I.; Flórez-Álvarez, L.; Yepes, L.M.; Rugeles, M.T.; Zapata-Builes, W.; Hernandez, J.C.; Taborda, N.A. Curcumin Inhibits In Vitro SARS-CoV-2 Infection In Vero E6 Cells through Multiple Antiviral Mechanisms. Molecules 2021, 26, 6900. [Google Scholar] [CrossRef]
- Di Pierro, F.; Derosa, G.; Maffioli, P.; Bertuccioli, A.; Togni, S.; Riva, A.; Allegrini, P.; Khan, A.; Khan, S.; Khan, B.A.; et al. Possible Therapeutic Effects of Adjuvant Quercetin Supplementation Against Early-Stage COVID-19 Infection: A Prospective, Randomized, Controlled, and Open-Label Study. Int. J. Gen. Med. 2021, 14, 2359–2366. [Google Scholar] [CrossRef]
- Jo, S.; Kim, S.; Shin, D.H.; Kim, M.S. Inhibition of SARS-CoV 3CL protease by flavonoids. J. Enzym. Inhib. Med. Chem. 2020, 35, 145–151. [Google Scholar] [CrossRef]
- Milanović Ž, B.; Antonijević, M.R.; Amić, A.D.; Avdović, E.H.; Dimić, D.S.; Milenković, D.A.; Marković, Z.S. Inhibitory activity of quercetin, its metabolite, and standard antiviral drugs towards enzymes essential for SARS-CoV-2: The role of acid-base equilibria. RSC Adv. 2021, 11, 2838–2847. [Google Scholar] [CrossRef]
- Barhouchi, B.; Menacer, R.; Bouchkioua, S.; Mansour, A.; Belattar, N. Compounds from myrtle flowers as antibacterial agents and SARS-CoV-2 inhibitors: In-vitro and molecular docking studies. Arab. J. Plant Prot. 2023, 16, 104939. [Google Scholar] [CrossRef]
- Guimarães Santana, B.C.; de Almeida Marques, D.P.; dos Santos Freitas, A.; Ferreira, M.M.; de Sousa Lopes, D.; Bagno, F.F.; Guimarães da Fonseca, F.; dos Reis, J.G.A.C.; Oliveira Mendes, T.A.d.; Santos, J.L.d.; et al. Protease inhibitors from Theobroma cacao impair SARS-CoV-2 replication in vitro. Heliyon 2023, 9, e15860. [Google Scholar] [CrossRef] [PubMed]
- Hossain, A.; Rahman, M.E.; Rahman, M.S.; Nasirujjaman, K.; Matin, M.N.; Faruqe, M.O.; Rabbee, M.F. Identification of medicinal plant-based phytochemicals as a potential inhibitor for SARS-CoV-2 main protease (Mpro) using molecular docking and deep learning methods. Comput. Biol. Med. 2023, 157, 106785. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-N.; Zhu, G.-H.; Liu, W.; Xiong, Y.; Hu, Q.; Zhuang, X.-Y.; Jia, G.-H.; Zhang, W.-D.; Ge, G.-B. Discovery and characterization of the covalent SARS-CoV-2 3CLpro inhibitors from Ginkgo biloba extract via integrating chemoproteomic and biochemical approaches. Phytomedicine 2023, 114, 154796. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Al-Rehaily, A.J.; Ahmad, M.S.; Yousaf, M.; Nur-e-Alam, M.; Parvez, M.K.; Al-Dosari, M.S.; Noman, O.M.; Khan, S.I.; Khan, I.A. Chemical constituents from Oncocalyx glabratus and their biological activities. Phytochem. Lett. 2017, 20, 128–132. [Google Scholar] [CrossRef]
- Ahmed, S.; Al-Rehaily, A.J.; Ahmad, M.S.; Yousaf, M.; Nur-e-Alam, M.; Thomas, J.; Khan, S.I.; Khan, I.A. Cytotoxic and antiinflammatory activities of the chemical constituents isolated from Baccharoides schimperi DC. S. Afr. J. Bot. 2018, 114, 9–13. [Google Scholar] [CrossRef]
- Ahmed, S.; Mothana, R.; Yousaf, M.; Al-Rehaily, A. Activity guided isolation of chemical constituents from the biologically active methanol extract of Euphorbia schimperi C. Presl. Bull. Chem. Soc. Ethiop. 2017, 31, 471–479. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Hakami, A.R.; Bakheit, A.H.; Almehizia, A.A.; Ghazwani, M.Y. Selection of SARS-CoV-2 main protease inhibitor using structure-based virtual screening. Future Med. Chem. 2021, 14, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Sanachai, K.; Mahalapbutr, P.; Sanghiran Lee, V.; Rungrotmongkol, T.; Hannongbua, S. In Silico Elucidation of Potent Inhibitors and Rational Drug Design against SARS-CoV-2 Papain-like Protease. J. Phys. Chem. B 2021, 125, 13644–13656. [Google Scholar] [CrossRef] [PubMed]
- Bakheit, A.H.; Attwa, M.W.; Kadi, A.A.; Alkahtani, H.M. Structural Analysis and Reactivity Insights of (E)-Bromo-4-((4-((1-(4-chlorophenyl)ethylidene)amino)-5-phenyl-4H-1,2,4-triazol-3-yl)thio)-5-((2-isopropylcyclohexyl)oxy) Furan-2(5H)-one: A Combined Approach Using Single-Crystal X-ray Diffraction, Hirshfeld Surface Analysis, and Conceptual Density Functional Theory. Crystals 2023, 13, 1313. [Google Scholar]
- Hognon, C.; Marazzi, M.; García-Iriepa, C. Atomistic-Level Description of the Covalent Inhibition of SARS-CoV-2 Papain-like Protease. Int. J. Mol. Sci. 2022, 23, 5855. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.A.; Banerjee, S.; Ghosh, K.; Gayen, S.; Jha, T. Protease targeted COVID-19 drug discovery and its challenges: Insight into viral main protease (Mpro) and papain-like protease (PLpro) inhibitors. Biorg. Med. Chem. 2021, 29, 115860. [Google Scholar] [CrossRef]
- Shen, Z.; Ratia, K.; Cooper, L.; Kong, D.; Lee, H.; Kwon, Y.; Li, Y.; Alqarni, S.; Huang, F.; Dubrovskyi, O.; et al. Design of SARS-CoV-2 PLpro Inhibitors for COVID-19 Antiviral Therapy Leveraging Binding Cooperativity. J. Med. Chem. 2022, 65, 2940–2955. [Google Scholar] [CrossRef]
- Liu, Y.; Liang, C.; Xin, L.; Ren, X.; Tian, L.; Ju, X.; Li, H.; Wang, Y.; Zhao, Q.; Liu, H.; et al. The development of Coronavirus 3C-Like protease (3CLpro) inhibitors from 2010 to 2020. Eur. J. Med. Chem. 2020, 206, 112711. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Kusov, Y.; Hilgenfeld, R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antivir. Res. 2018, 149, 58–74. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Huang, Y.; Lau, S.K.P.; Yuen, K.-Y. Coronavirus Genomics and Bioinformatics Analysis. Viruses 2010, 2, 1804–1820. [Google Scholar] [CrossRef] [PubMed]
- Barretto, N.; Jukneliene, D.; Ratia, K.; Chen, Z.; Mesecar Andrew, D.; Baker Susan, C. The Papain-Like Protease of Severe Acute Respiratory Syndrome Coronavirus Has Deubiquitinating Activity. J. Virol. 2005, 79, 15189–15198. [Google Scholar] [CrossRef] [PubMed]
- Báez-Santos, Y.M.; St. John, S.E.; Mesecar, A.D. The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antivir. Res. 2015, 115, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Osipiuk, J.; Azizi, S.-A.; Dvorkin, S.; Endres, M.; Jedrzejczak, R.; Jones, K.A.; Kang, S.; Kathayat, R.S.; Kim, Y.; Lisnyak, V.G.; et al. Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat. Commun. 2021, 12, 743. [Google Scholar] [CrossRef] [PubMed]
- Csermely, P.; Ágoston, V.; Pongor, S. The efficiency of multi-target drugs: The network approach might help drug design. Trends Pharmacol. Sci. 2005, 26, 178–182. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings1PII of original article: S0169-409X(96)00423-1. The article was originally published in Adv. Drug Deliv. Rev. 1997, 23, 3–25. Adv. Drug Del. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Padhi, S.; Masi, M.; Chourasia, R.; Rajashekar, Y.; Rai, A.K.; Evidente, A. ADMET profile and virtual screening of plant and microbial natural metabolites as SARS-CoV-2 S1 glycoprotein receptor binding domain and main protease inhibitors. Eur. J. Pharmacol. 2021, 890, 173648. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Sultana, I.; Shuvo, M.N.; Shil, A.; Himel, M.K.; Hasan, M.A.; Shawan, M. In Silico Identification and Analysis of Potentially Bioactive Antiviral Phytochemicals against SARS-CoV-2: A Molecular Docking and Dynamics Simulation Approach. BioMed Res. Int. 2023, 2023, 5469258. [Google Scholar] [CrossRef] [PubMed]
- Khanal, P.; Patil, V.S.; Bhandare, V.V.; Dwivedi, P.S.R.; Shastry, C.S.; Patil, B.M.; Gurav, S.S.; Harish, D.R.; Roy, S. Computational investigation of benzalacetophenone derivatives against SARS-CoV-2 as potential multi-target bioactive compounds. Comput. Biol. Med. 2022, 146, 105668. [Google Scholar] [CrossRef] [PubMed]
- Mosquera-Yuqui, F.; Lopez-Guerra, N.; Moncayo-Palacio, E.A. Targeting the 3CLpro and RdRp of SARS-CoV-2 with phytochemicals from medicinal plants of the Andean Region: Molecular docking and molecular dynamics simulations. J. Biomol. Struct. Dyn. 2022, 40, 2010–2023. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Li, Y.; Xia, Y.-L.; Ai, S.-M.; Liang, J.; Sang, P.; Ji, X.-L.; Liu, S.-Q. Insights into Protein–Ligand Interactions: Mechanisms, Models, and Methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef]
- Koulgi, S.; Jani, V.; Uppuladinne, V.N.M.; Sonavane, U.; Joshi, R. Natural plant products as potential inhibitors of RNA dependent RNA polymerase of Severe Acute Respiratory Syndrome Coronavirus-2. PLoS ONE 2021, 16, e0251801. [Google Scholar] [CrossRef]
- Kawall, A.; Lewis, D.S.M.; Sharma, A.; Chavada, K.; Deshmukh, R.; Rayalam, S.; Mody, V.; Taval, S. Inhibitory effect of phytochemicals towards SARS-CoV-2 papain like protease (PLpro) proteolytic and deubiquitinase activity. Front. Chem. 2023, 10, 1100460. [Google Scholar] [CrossRef]
- Pillaiyar, T.; Manickam, M.; Namasivayam, V.; Hayashi, Y.; Jung, S.-H. An Overview of Severe Acute Respiratory Syndrome–Coronavirus (SARS-CoV) 3CL Protease Inhibitors: Peptidomimetics and Small Molecule Chemotherapy. J. Med. Chem. 2016, 59, 6595–6628. [Google Scholar] [CrossRef]
- Tang, B.; He, F.; Liu, D.; He, F.; Wu, T.; Fang, M.; Niu, Z.; Wu, Z.; Xu, D. AI-Aided Design of Novel Targeted Covalent Inhibitors against SARS-CoV-2. Biomolecules 2022, 12, 746. [Google Scholar] [CrossRef]
- Gehringer, M.; Laufer, S.A. Emerging and Re-Emerging Warheads for Targeted Covalent Inhibitors: Applications in Medicinal Chemistry and Chemical Biology. J. Med. Chem. 2019, 62, 5673–5724. [Google Scholar] [CrossRef]
- Huang, F.; Han, X.; Xiao, X.; Zhou, J. Covalent Warheads Targeting Cysteine Residue: The Promising Approach in Drug Development. Molecules 2022, 27, 7728. [Google Scholar] [CrossRef]
- Liang, S.T.; Chen, C.; Chen, R.X.; Li, R.; Chen, W.L.; Jiang, G.H.; Du, L.L. Michael acceptor molecules in natural products and their mechanism of action. Front. Pharmacol. 2022, 13, 1033003. [Google Scholar] [CrossRef]
- Zmudzinski, M.; Rut, W.; Olech, K.; Granda, J.; Giurg, M.; Burda-Grabowska, M.; Kaleta, R.; Zgarbova, M.; Kasprzyk, R.; Zhang, L.; et al. Ebselen derivatives inhibit SARS-CoV-2 replication by inhibition of its essential proteins: PLpro and Mpro proteases, and nsp14 guanine N7-methyltransferase. Sci. Rep. 2023, 13, 9161. [Google Scholar] [CrossRef]
- Rut, W.; Lv, Z.; Zmudzinski, M.; Patchett, S.; Nayak, D.; Snipas, S.J.; El Oualid, F.; Huang, T.T.; Bekes, M.; Drag, M.; et al. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: A framework for anti–COVID-19 drug design. Sci. Adv. 2020, 6, eabd4596. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Qin, B.; Chen, P.; Zhu, K.; Hou, P.; Wojdyla, J.A.; Wang, M.; Cui, S. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sin. B 2021, 11, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Klemm, T.; Ebert, G.; Calleja, D.J.; Allison, C.C.; Richardson, L.W.; Bernardini, J.P.; Lu, B.G.C.; Kuchel, N.W.; Grohmann, C.; Shibata, Y.; et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020, 39, e106275. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, J.; Polshakov, D.; Kato-Weinstein, J.; Zhou, Q.; Li, Y.; Granet, R.; Garner, L.; Deng, Y.; Liu, C.; Albaiu, D.; et al. Quantitative Structure–Activity Relationship Machine Learning Models and their Applications for Identifying Viral 3CLpro- and RdRp-Targeting Compounds as Potential Therapeutics for COVID-19 and Related Viral Infections. ACS Omega 2020, 5, 27344–27358. [Google Scholar] [CrossRef] [PubMed]
- Welker, A.; Kersten, C.; Müller, C.; Madhugiri, R.; Zimmer, C.; Müller, P.; Zimmermann, R.; Hammerschmidt, S.; Maus, H.; Ziebuhr, J.; et al. Structure-Activity Relationships of Benzamides and Isoindolines Designed as SARS-CoV Protease Inhibitors Effective against SARS-CoV-2. ChemMedChem 2021, 16, 340–354. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Roy, K. Development of a simple, interpretable and easily transferable QSAR model for quick screening antiviral databases in search of novel 3C-like protease (3CLpro) enzyme inhibitors against SARS-CoV diseases. SAR QSAR Environ. Res. 2020, 31, 511–526. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Dai, R.; Su, R. Computer-aided drug design for the pain-like protease (PLpro) inhibitors against SARS-CoV-2. Biomed. Pharmacother. 2023, 159, 114247. [Google Scholar] [CrossRef] [PubMed]
- Pitera, J.W. Expected Distributions of Root-Mean-Square Positional Deviations in Proteins. J. Phys. Chem. B 2014, 118, 6526–6530. [Google Scholar] [CrossRef]
- Azeem, M.; Mustafa, G.; Mahrosh, H.S. Virtual screening of phytochemicals by targeting multiple proteins of severe acute respiratory syndrome coronavirus 2: Molecular docking and molecular dynamics simulation studies. Int. J. Immunopathol. Pharmacol. 2022, 36, 03946320221142793. [Google Scholar] [CrossRef]
- Bornot, A.; Etchebest, C.; de Brevern, A.G. Predicting protein flexibility through the prediction of local structures. Proteins Struct. Funct. Bioinform. 2011, 79, 839–852. [Google Scholar] [CrossRef]
- Al-Otaibi, J.S.; Sheena Mary, Y.; Shyma Mary, Y.; Acharjee, N.; Balachandar, S.; Yathirajan, H.S. Insights into solvation effects, spectroscopic, Hirshfeld surface Analysis, reactivity analysis and anti-Covid-19 ability of doxylamine succinate: Experimental, DFT, MD and docking simulations. J. Mol. Liq. 2022, 361, 119609. [Google Scholar] [CrossRef]
- Ghazwani, M.Y.; Bakheit, A.H.; Hakami, A.R.; Alkahtani, H.M.; Almehizia, A.A. Virtual Screening and Molecular Docking Studies for Discovery of Potential RNA-Dependent RNA Polymerase Inhibitors. Crystals 2021, 11, 471. [Google Scholar] [CrossRef]
- Weng, Y.L.; Naik, S.R.; Dingelstad, N.; Lugo, M.R.; Kalyaanamoorthy, S.; Ganesan, A. Molecular dynamics and in silico mutagenesis on the reversible inhibitor-bound SARS-CoV-2 main protease complexes reveal the role of lateral pocket in enhancing the ligand affinity. Sci. Rep. 2021, 11, 7429. [Google Scholar] [CrossRef] [PubMed]
- Parihar, A.; Sonia, Z.F.; Akter, F.; Ali, M.A.; Hakim, F.T.; Hossain, M.S. Phytochemicals-based targeting RdRp and main protease of SARS-CoV-2 using docking and steered molecular dynamic simulation: A promising therapeutic approach for Tackling COVID-19. Comput. Biol. Med. 2022, 145, 105468. [Google Scholar] [CrossRef] [PubMed]
- ChemAxon. Available online: https://chemaxon.com/marvin (accessed on 3 September 2023).
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- ProTox-II—Prediction of Toxicity of Chemicals. Available online: https://tox-new.charite.de/protox_II/ (accessed on 3 September 2023).
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Mesecar, A.D. Structure of COVID-19 main protease bound to potent broad-spectrum non-covalent inhibitor X77. Protein Data Bank. 2020. [Google Scholar] [CrossRef]
- Khalaf, H.S.; Naglah, A.M.; Al-Omar, M.A.; Moustafa, G.O.; Awad, H.M.; Bakheit, A.H. Synthesis, Docking, Computational Studies, and Antimicrobial Evaluations of New Dipeptide Derivatives Based on Nicotinoylglycylglycine Hydrazide. Molecules 2020, 25, 3589. [Google Scholar] [CrossRef]
- Al-Khodairy, F.M.; Khan, M.K.A.; Kunhi, M.; Pulicat, M.S.; Akhtar, S.; Arif, J.M. In Silico prediction of mechanism of Erysolin-induced apoptosis in human breast cancer cell lines. Am. J. Bioinform. Res. 2013, 3, 62–71. [Google Scholar]
- Labute, P. Protonate3D: Assignment of ionization states and hydrogen coordinates to macromolecular structures. Proteins: Struct. Funct. Bioinform. 2009, 75, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, A.; Wan, X.; Burlingame, A.L.; Taunton, J. Ligand Conformational Bias Drives Enantioselective Modification of a Surface-Exposed Lysine on Hsp90. J. Am. Chem. Soc. 2020, 142, 3392–3400. [Google Scholar] [CrossRef] [PubMed]
- Wojciechowski, M.; Lesyng, B. Generalized Born Model: Analysis, Refinement, and Applications to Proteins. J. Phys. Chem. B 2004, 108, 18368–18376. [Google Scholar] [CrossRef]
- Corbeil, C.R.; Williams, C.I.; Labute, P. Variability in docking success rates due to dataset preparation. J. Comput.-Aided Mol. Des. 2012, 26, 775–786. [Google Scholar] [CrossRef] [PubMed]
- Naïm, M.; Bhat, S.; Rankin, K.N.; Dennis, S.; Chowdhury, S.F.; Siddiqi, I.; Drabik, P.; Sulea, T.; Bayly, C.I.; Jakalian, A.; et al. Solvated Interaction Energy (SIE) for Scoring Protein−Ligand Binding Affinities. 1. Exploring the Parameter Space. J. Chem. Inf. Model. 2007, 47, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Ghufran, M.; Ullah, M.; Khan, H.A.; Ghufran, S.; Ayaz, M.; Siddiq, M.; Abbas, S.Q.; Hassan, S.S.; Bungau, S. In-Silico Lead Druggable Compounds Identification against SARS COVID-19 Main Protease Target from In-House, Chembridge and Zinc Databases by Structure-Based Virtual Screening, Molecular Docking and Molecular Dynamics Simulations. Bioengineering 2023, 10, 100. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Lee, J.; Smith, I.P.S.; Lee, H.; Kim, S.; Qi, Y.; Klauda, J.B.; Widmalm, G.; Khalid, S.; Im, W. CHARMM-GUI Supports Hydrogen Mass Repartitioning and Different Protonation States of Phosphates in Lipopolysaccharides. J. Chem. Inf. Model. 2021, 61, 831–839. [Google Scholar] [CrossRef] [PubMed]
- CHARMM General Force Field (CGenFF). Available online: https://cgenff.silcsbio.com (accessed on 3 September 2023).
- Yu, W.; He, X.; Vanommeslaeghe, K.; MacKerell, A.D., Jr. Extension of the CHARMM general force field to sulfonyl-containing compounds and its utility in biomolecular simulations. J. Comput. Chem. 2012, 33, 2451–2468. [Google Scholar] [CrossRef]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, S.; Onck, P.R.; Giessen, E.v.d. CHARMM TIP3P Water Model Suppresses Peptide Folding by Solvating the Unfolded State. J. Phys. Chem. B 2016, 120, 3692–3698. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Z.; Luginsland, J.; Thomas, R.J.; Dennis, P.B.; Kelley-Loughnane, N.; Roach, W.P.; Naik, R.R. Molecular dynamics simulations explore effects of electric field orientations on spike proteins of SARS-CoV-2 virions. Sci. Rep. 2022, 12, 12986. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Code | Compound Name | 2D Structure | Name of Plants |
|---|---|---|---|
| 1 | 5,3′,4′-trihydroxyflavan 7-O-gallate |  | Oncocalyx glabratus |
| 2 | 5,4′-dihydroxyflavan 7-3′-O-digallate |  | Oncocalyx glabratus |
| 3 | 5,3′-dihydroxyflavan 7-4′-O-digallate |  | Oncocalyx glabratus |
| 4 | Spinasterol |  | Baccharoides schimperi |
| 5 | Stigmasterol |  | Baccharoides schimperi |
| 6 | 3′,4′,5,7-tetrahydroxy-3-methoxyflavone |  | Baccharoides schimperi |
| 7 | Vernolepin |  | Baccharoides schimperi |
| 8 | Vernodalol |  | Baccharoides schimperi |
| 9 | 11β,13-dihydrovernodalin |  | Baccharoides schimperi |
| 10 | Quercitrin 3-O-rhamnoside |  | Euphorpia schimperi |
| Compound Code | A LogP | Mol. Wt. | nHBA | nHBD | MFPSA | Num-Ring | No. R. Bonds | TPSA | MR |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 4.178 | 410.374 | 8 | 5 | 0.364 | 4 | 4 | 136.68 | 105.42 |
| 2 | 5.126 | 562.478 | 12 | 7 | 0.401 | 5 | 7 | 203.44 | 140.87 |
| 3 | 5.126 | 562.478 | 12 | 7 | 0.401 | 5 | 7 | 203.44 | 140.87 |
| 4 | 7.639 | 412.691 | 1 | 1 | 0.042 | 4 | 5 | 20.23 | 128.69 |
| 5 | 7.639 | 412.691 | 1 | 1 | 0.042 | 4 | 5 | 20.23 | 128.69 |
| 6 | 1.856 | 316.262 | 7 | 4 | 0.397 | 3 | 2 | 120.36 | 82.50 |
| 7 | 0.818 | 276.285 | 5 | 1 | 0.273 | 3 | 1 | 72.83 | 66.87 |
| 8 | 0.892 | 392.4 | 8 | 2 | 0.291 | 2 | 8 | 119.36 | 95.00 |
| 9 | 1.236 | 362.374 | 7 | 1 | 0.275 | 3 | 5 | 99.13 | 86.88 |
| 10 | 0.589 | 448.377 | 11 | 7 | 0.462 | 4 | 3 | 190.28 | 103.90 |
| Compound Code | HIA Level | CYP2D6 | Hepatotoxicity | PPB | Solubility Level | Molar Solubility Log(sw) | BBBP Level | AlogP98 | PSA_2D |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | True | False | False | 2 | −5.103 | 4 | 4.178 | 139.238 |
| 2 | 3 | True | False | False | 1 | −7.249 | 4 | 5.126 | 207.1 |
| 3 | 3 | True | False | False | 1 | −7.127 | 4 | 5.126 | 207.1 |
| 4 | 3 | True | False | True | 1 | −7.962 | 4 | 7.639 | 20.815 |
| 5 | 3 | False | False | True | 1 | −7.963 | 4 | 7.639 | 20.815 |
| 6 | 0 | True | False | False | 3 | −2.93 | 4 | 1.856 | 118.422 |
| 7 | 0 | False | False | True | 3 | −2.392 | 3 | 0.818 | 73.277 |
| 8 | 0 | False | False | True | 3 | −2.065 | 4 | 0.893 | 120.323 |
| 9 | 0 | False | False | True | 3 | −2.842 | 3 | 1.236 | 99.508 |
| 10 | 3 | False | False | False | 3 | −3.888 | 4 | 0.589 | 189.799 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saquib, Q.; Bakheit, A.H.; Ahmed, S.; Ansari, S.M.; Al-Salem, A.M.; Al-Khedhairy, A.A. Identification of Phytochemicals from Arabian Peninsula Medicinal Plants as Strong Binders to SARS-CoV-2 Proteases (3CLPro and PLPro) by Molecular Docking and Dynamic Simulation Studies. Molecules 2024, 29, 998. https://doi.org/10.3390/molecules29050998
Saquib Q, Bakheit AH, Ahmed S, Ansari SM, Al-Salem AM, Al-Khedhairy AA. Identification of Phytochemicals from Arabian Peninsula Medicinal Plants as Strong Binders to SARS-CoV-2 Proteases (3CLPro and PLPro) by Molecular Docking and Dynamic Simulation Studies. Molecules. 2024; 29(5):998. https://doi.org/10.3390/molecules29050998
Chicago/Turabian StyleSaquib, Quaiser, Ahmed H. Bakheit, Sarfaraz Ahmed, Sabiha M. Ansari, Abdullah M. Al-Salem, and Abdulaziz A. Al-Khedhairy. 2024. "Identification of Phytochemicals from Arabian Peninsula Medicinal Plants as Strong Binders to SARS-CoV-2 Proteases (3CLPro and PLPro) by Molecular Docking and Dynamic Simulation Studies" Molecules 29, no. 5: 998. https://doi.org/10.3390/molecules29050998
APA StyleSaquib, Q., Bakheit, A. H., Ahmed, S., Ansari, S. M., Al-Salem, A. M., & Al-Khedhairy, A. A. (2024). Identification of Phytochemicals from Arabian Peninsula Medicinal Plants as Strong Binders to SARS-CoV-2 Proteases (3CLPro and PLPro) by Molecular Docking and Dynamic Simulation Studies. Molecules, 29(5), 998. https://doi.org/10.3390/molecules29050998