

A Sustainable Synthetic Approach to Tacrine and Cholinesterase Inhibitors in Deep Eutectic Solvents under Aerobic Conditions




Abstract
:1. Introduction
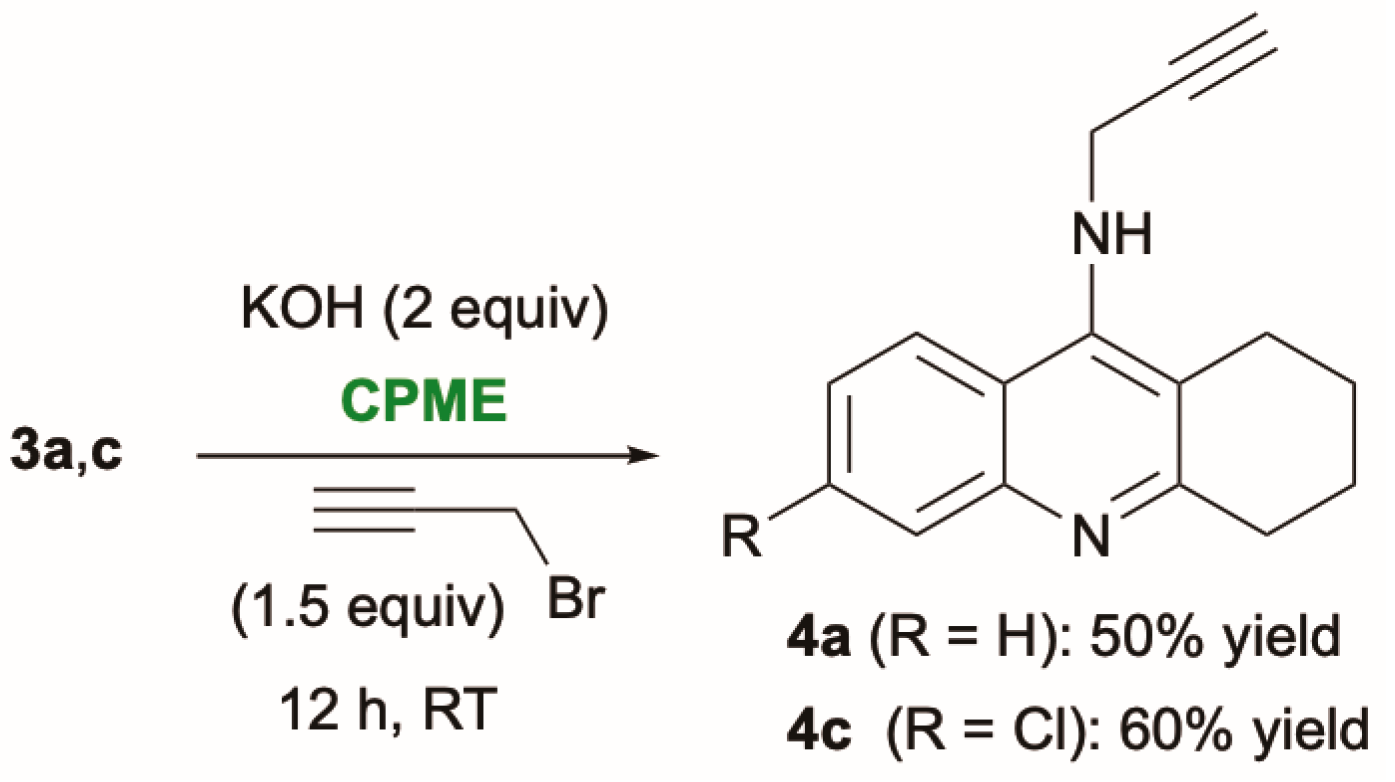
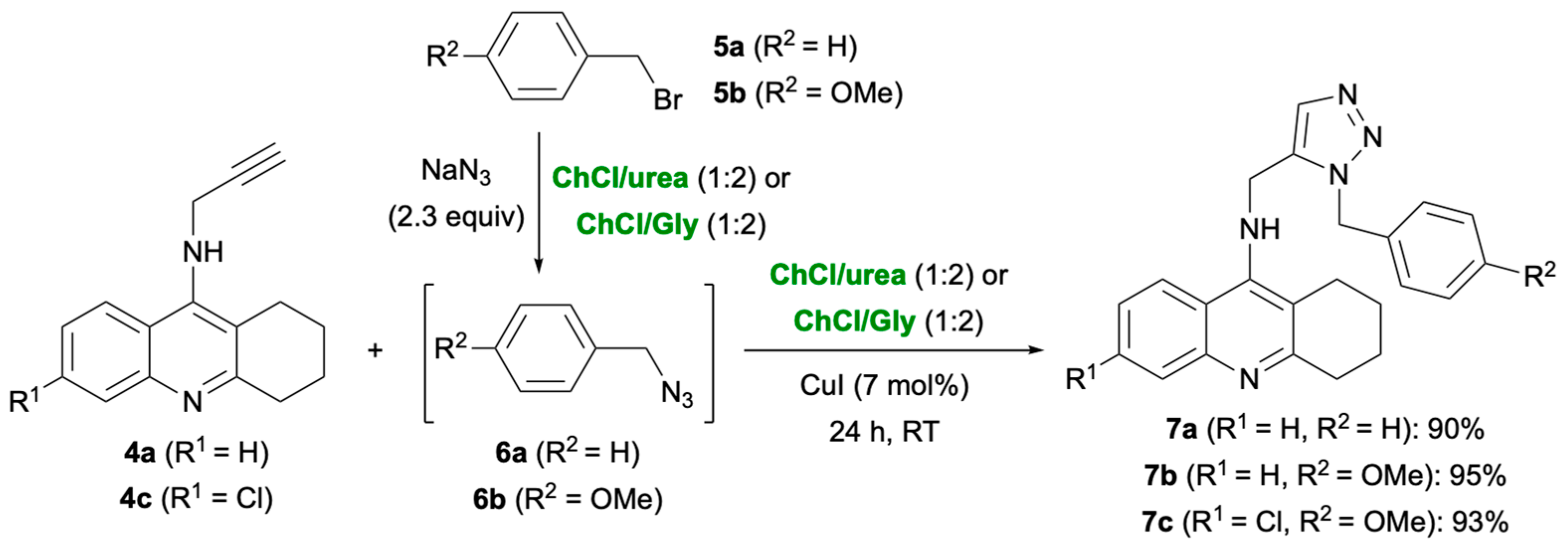
2. Results and Discussion
3. Materials and Methods
3.1. General Methods
3.2. Synthesis of Tacrine 3a in ZnCl2/ChCl (1:1 mol mol−1) LADES
3.3. Synthesis of Tacrine Analogue 3c in ZnCl2/ChCl (1:1 mol mol−1) or FeCl3·6H2O/urea (2:1 mol mol−1) LADES
3.4. Scale-Up Synthesis of Tacrine 3a in ZnCl2/ChCl (1:1 mol mol−1) LADES
3.5. Representative Procedure for the Synthesis of N-Propargyl-Substituted Tacrine Derivatives 4a,c in CPME: Synthesis of 4a
3.6. Representative Procedure for the Synthesis of Triazole-Based Tacrine Derivatives 7a–c: Synthesis of 7a
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sheldon, R.A. Green solvents for sustainable organic synthesis: State of the art. Green Chem. 2005, 7, 267–278. [Google Scholar] [CrossRef]
- Lipshutz, B.H.; Gallou, F.; Handa, S. Evolution of Solvents in Organic Synthesis. ACS Sustain. Chem. Eng. 2016, 4, 5838–5849. [Google Scholar] [CrossRef]
- Sanches, M.V.; Freitas, R.; Oliva, M.; Mero, A.; De Marchi, L.; Cuccaro, A.; Fumagalli, G.; Mezzetta, A.; Dugoni, G.C.; Ferro, M.; et al. Are natural deep eutectic solvents always a sustainable option? A bioassay-based study. Environ. Sci. Pollut. Res. 2023, 30, 17268–17279. [Google Scholar] [CrossRef]
- Lomba, L.; Ribate, M.P.; Sangüesa, E.; Concha, J.; Garralaga, M.P.; Errazquin, D.; García, C.B.; Giner, B. Deep Eutectic Solvents: Are They Safe? Appl. Sci. 2021, 11, 10061. [Google Scholar] [CrossRef]
- Martínez Martínez, G.; Guillena Townley, G. Controversy on the toxic nature of deep eutectic solvents and their potential contribution to environmental pollution. Heliyon 2022, 8, e12567. [Google Scholar] [CrossRef]
- Zainal-Abidin, M.H.; Hayyan, M.; Ngoh, G.C.; Wong, W.F.; Looi, C.Y. Emerging frontiers of deep eutectic solvents in drug discovery and drug delivery systems. J. Control. Release 2019, 316, 168–195. [Google Scholar] [PubMed]
- Hansen, B.B.; Spittle, S.; Chen, B.; Poe, D.; Zhang, Y.; Klein, J.M.; Horton, A.; Adhikari, L.; Zelovich, T.; Doherty, B.W.; et al. Deep Eutectic Solvents: A Review of Fundamentals and Applications. Chem. Rev. 2021, 121, 1232–1285. [Google Scholar] [PubMed]
- Majee, S.; Shilpa; Sarav, M.; Banik, B.K.; Ray, D. Recent Advances in the Green Synthesis of Active N-Heterocycles and Their Biological Activities. Pharmaceuticals 2023, 16, 873. [Google Scholar] [CrossRef]
- Perna, F.M.; Vitale, P.; Capriati, V. Organic Synthesis in DESs in Deep Eutectic Solvents: Synthesis, Properties, and Applications, 1st ed.; Ramón, D.J., Guillena, G., Eds.; Wiley-VCH Verlag GmbH & Co: Weinheim, Germany, 2020; Volume 7, pp. 111–134. [Google Scholar]
- Gore, S.; Baskaran, S.; König, B. Fischer Indole Synthesis in Low Melting Mixtures. Org. Lett. 2012, 14, 4568–4571. [Google Scholar] [CrossRef]
- Shaibuna, M.; Sreekumar, K. Dual solvent-catalyst role of deep eutectic solvents in Hantzsch dihydropyridine synthesis. Synth. Commun. 2021, 51, 1742–1753. [Google Scholar]
- Alvi, S.; Ali, R. An expeditious and highly efficient synthesis of substituted pyrroles using a low melting deep eutectic mixture. Org. Biomol. Chem. 2021, 19, 9732–9745. [Google Scholar] [CrossRef]
- Vitale, P.; Cicco, L.; Cellamare, I.; Perna, F.M.; Salomone, A.; Capriati, V. Regiodivergent synthesis of functionalized pyrimidines and imidazoles through phenacyl azides in deep eutectic solvents. Beilstein J. Org. Chem. 2020, 16, 1915–1923. [Google Scholar] [CrossRef]
- Capua, M.; Perrone, S.; Perna, F.M.; Vitale, P.; Troisi, L.; Salomone, S.; Capriati, V. An Expeditious and Greener Synthesis of 2-Aminoimidazoles in Deep Eutectic Solvents. Molecules 2016, 21, 924. [Google Scholar] [CrossRef]
- Perrone, S.; Capua, M.; Messa, F.; Salomone, A.; Troisi, L. Green synthesis of 2-pyrazinones in deep eutectic solvents: From α-chloro oximes to peptidomimetic scaffolds. Tetrahedron 2017, 73, 6193–6198. [Google Scholar] [CrossRef]
- Behalo, M.S.; Bloise, E.; Mele, G.; Salomone, A.; Messa, F.; Carbone, L.; Mazzetto, S.E.; Lomonaco, D. Bio-based benzoxazines synthesized in a deep eutectic solvent: A greener approach toward vesicular nanosystems. J. Heterocycl. Chem. 2020, 57, 768–773. [Google Scholar] [CrossRef]
- Dilauro, G.; Cicco, L.; Vitale, P.; Perna, F.M.; Capriati, V. Ligand-Free Pd-Catalyzed Reductive Mizoroki-Heck Reaction Strategy for the One-Pot Synthesis of Functionalized Oxygen Heterocycles in Deep Eutectic Solvents. Eur. J. Org. Chem. 2023, 26, e202200814. [Google Scholar] [CrossRef]
- Cicco, L.; Perna, F.M.; Falcicchio, A.; Altomare, A.; Messa, F.; Salomone, A.; Capriati, V.; Vitale, P. 1,3-Dipolar Cycloaddition of Alkanone Enolates with Azides in Deep Eutectic Solvents for the Metal-Free Regioselective Synthesis of Densely Functionalized 1,2,3-Triazoles. Eur. J. Org. Chem. 2022, 36, e202200843. [Google Scholar] [CrossRef]
- Cicco, L.; Hernández-Fernández, J.A.; Salomone, A.; Vitale, P.; Ramos-Martín, M.; González-Sabín, J.; Presa Soto, A.; Perna, F.M.; Capriati, V.; García-Álvarez, J. Copper-catalyzed Goldberg-type C-N coupling in deep eutectic solvents (DESs) and water under aerobic conditions. Org. Biomol. Chem. 2021, 19, 1773–1779. [Google Scholar] [CrossRef]
- Quivelli, A.F.; Rossi, F.V.; Vitale, P.; García-Álvarez, J.; Perna, F.M.; Capriati, V. Sustainable and Scalable Two-Step Synthesis of Thenfadil and Some Analogs in Deep Eutectic Solvents: From Laboratory to Industry. ACS Sustain. Chem. Eng. 2022, 10, 4065–4072. [Google Scholar] [CrossRef]
- Quivelli, A.F.; Rossi, F.V.; Alario, C.; Sannicolò, F.; Vitale, P.; García-Álvarez, J.; Perna, F.M.; Capriati, V. Green Solvents for Eco-Friendly Synthesis of Dimethindene: A Forward-Looking Approach. Molecules 2022, 27, 7594. [Google Scholar] [CrossRef]
- Janočková, J.; Plšíková, J.; Koval’, J.; Jendželovský, R.; Mikeš, R.; Kašpárková, J.; Brabec, V.; Hamuľaková, S.; Fedoročko, P.; Kožurková, M. Tacrine derivatives as dual topoisomerase I and II catalytic inhibitors. Bioorg. Chem. 2015, 59, 168–176. [Google Scholar] [CrossRef]
- Gulati, H.K.; Choudhary, S.; Kumar, N.; Ahmed, A.; Bhagat, K.; Singh, J.V.; Singh, A.; Kumar, A.; Bedi, P.M.S.; Singh, H. Design, Synthesis, biological investigations and molecular interactions of triazole linked tacrine glycoconjugates as Acetylcholinesterase inhibitors with reduced hepatotoxicity. Bioorg. Chem. 2022, 118, 105479. [Google Scholar]
- Maspero, M.; Volpato, D.; Cirillo, D.; Chen, N.Y.; Messerer, R.; Sotriffer, C.; De Amici, M.; Holzgrabe, U.; Dallanoce, C. Tacrine-xanomeline and tacrine-iperoxo hybrid ligands: Synthesis and biological evaluation at acetylcholinesterase and M1 muscarinic acetylcholine receptors. Bioorg. Chem. 2020, 96, 103633. [Google Scholar] [CrossRef]
- Tang, J.; Li, J.; Zhang, L.; Ma, S.; Shi, D.; Zhang, Q.; Yang, L.; Wang, X.; Liu, X.; Liu, C. The Divergent Transformations of Aromatic o-Aminonitrile with Carbonyl Compound. J. Heterocycl. Chem. 2012, 49, 533–542. [Google Scholar] [CrossRef]
- Qin, Q.-P.; Wang, S.-L.; Tan, M.-X.; Wang, Z.-F.; Luo, D.-M.; Zou, B.-Q.; Liu, Y.-C. Novel tacrine platinum(II) complexes display high anticancer activity via inhibition of telomerase activity, dysfunction of mitochondria, and activation of the p53 signaling pathway. Eur. J. Med. Chem. 2018, 158, 106–122. [Google Scholar] [CrossRef] [PubMed]
- Broichhagen, J.; Jurastow, I.; Iwan, K.; Kummer, W.; Trauner, D. Optical Control of Acetylcholinesterase with a Tacrine switch. Angew. Chem. Int. Ed. 2014, 53, 7657–7660. [Google Scholar] [CrossRef] [PubMed]
- Khalilzadeh, M.A.; Hosseini, A.; Tajbakhsh, M. Synthesis of Tacrine Derivatives Under Solventless Conditions. J. Heterocyclic Chem. 2007, 44, 535–538. [Google Scholar] [CrossRef]
- Fernández-Bachiller, M.I.; Pérez, C.; Campillo, N.E.; Páez, J.A.; González-Muñoz, G.C.; Usan, P.; García-Palomero, E.; López, M.G.; Villarroya, M.; García, A.G.; et al. Tacrine–Melatonin Hybrids as Multifunctional Agents for Alzheimer’s Disease, with Cholinergic, Antioxidant, and Neuroprotective Properties. ChemMedChem 2009, 4, 828–841. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Bachiller, M.I.; Pérez, C.; González-Munoz, G.C.; Conde, S.; López, M.G.; Villarroya, M.; García, A.G.; Rodríguez-Franco, M.I. Novel Tacrine–8-hydroxyquinoline hybrids as multifunctional agents for the treatment of alzheimer’s disease, with neuroprotective, cholinergic, antioxidant, and copper-complexing properties. J. Med. Chem. 2010, 53, 4927–4937. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Bachiller, M.I.; Pérez, C.; Monjas, L.; Rademann, J.; Rodríguez-Franco, M.I. New tacrine-4-oxo-4H-chromene hybrids as multifunctional agents for the treatment of Alzheimer’s disease, with cholinergic, antioxidant, and β-amyloid-reducing properties. J. Med. Chem. 2012, 55, 1303–1317. [Google Scholar] [CrossRef]
- Xie, S.-S.; Lan, J.-S.; Wang, X.-B.; Jiang, N.; Dong, G.; Li, Z.-R.; Wang, K.D.; Guo, P.-P.; Kong, L.-Y. Multifunctional tacrine– trolox hybrids for the treatment of Alzheimer’s disease with cholinergic, antioxidant, neuroprotective and hepatoprotective properties. Eur. J. Med. Chem. 2015, 93, 42–50. [Google Scholar] [CrossRef]
- Xie, S.; Chen, J.; Li, X.; Su, T.; Wang, Y.; Wang, Z.; Huang, L.; Li, X. Synthesis and evaluation of selegiline derivatives as monoamine oxidase inhibitor, antioxidant and metal chelator against Alzheimer’s disease. Biorg. Med. Chem. 2015, 23, 3722–3729. [Google Scholar] [CrossRef]
- McElroy, C.R.; Constantinou, A.; Jones, L.C.; Summerton, L.; Clark, J.H. Towards a holistic approach to metrics for the 21th century pharmaceutical industry. Green Chem. 2015, 17, 3111–3121. [Google Scholar] [CrossRef]
- Hu, H.; Song, L.; Fang, Q.; Zheng, J.; Meng, Z.; Luo, Y. Tin (IV) chloride-promoted one-pot synthesis of novel tacrine analogues. Molecules 2011, 22, 1878–1887. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Hu, X.; Wang, J.; Cheng, H.; Chen, L.; Qi, Z. Overview of the acidic deep eutectic solvents on synthesis, properties and applications. Green Energy Environ. 2020, 5, 8–21. [Google Scholar] [CrossRef]
- Mao, F.; Li, J.; Wei, H.; Huang, L.; Li, X. Tacrine–propargylamine derivatives with improved acetylcholinesterase inhibitory activity and lower hepatotoxicity as a potential lead compound for the treatment of Alzheimer’s disease. J. Enzyme Inhib. Med. Chem. 2015, 30, 995–1001. [Google Scholar] [CrossRef]
- Chrienova, Z.; Nepovimova, E.; Andrys, R.; Dolezal, R.; Janockova, J.; Muchova, L.; Fabova, L.; Soukup, O.; Oleksak, P.; Korabecny, J.; et al. Privileged multi-target directed propargyl-tacrines combining cholinesterase and monoamine oxidase inhibition activities. J. Enzyme Inhib. Med. Chem. 2022, 37, 2605–2620. [Google Scholar] [CrossRef] [PubMed]
- Azzena, U.; Carraro, M.; Pisano, L.; Monticelli, S.; Bartolotta, R.; Pace, V. Cyclopentyl Methyl Ether: An Elective Ecofriendly Ethereal Solvent in Classical and Modern Organic Chemistry. ChemSusChem 2019, 12, 40–70. [Google Scholar] [CrossRef]
- Vitale, P.; Lavolpe, F.; Valerio, F.; Di Biase, M.; Perna, F.M.; Messina, E.; Agrimi, G.; Pisano, I.; Capriati, V. Sustainable chemo-enzymatic preparation of enantiopure (R)-β-hydroxy-1,2,3-triazoles via lactic acid bacteria-mediated bioreduction of aromatic ketones and a heterogeneous “click” cycloaddition reaction in deep eutectic solvents. React. Chem. Eng. 2020, 5, 859–864. [Google Scholar] [CrossRef]
- Rastegari, A.; Safavi, M.; Vafadarnejad, F.; Najafi, Z.; Hariri, R.; Bukhari, S.N.A.; Iraji, A.; Edraki, N.; Firuzi, O.; Saeedi, M.; et al. Synthesis and evaluation of novel arylisoxazoles linked to tacrine moiety: In vitro and in vivo biological activities against Alzheimer’s disease. Mol. Divers. 2022, 26, 409–428. [Google Scholar] [CrossRef]
- Sheldon, R.A.; Bode, M.L.; Akakios, S.G. Metrics of green chemistry: Waste minimization. Curr. Opin. Green Sustain. Chem. 2022, 33, 100569. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||
|---|---|---|
| Entry | Acidic Deep Eutectic Solvents (ADESs) | Yield 3a (%) b |
| 1 | ChCl/Gly (1:2 mol mol−1) + ZnCl2 (20 mol%) | 20 |
| 2 | ChCl/urea (1:2 mol mol−1) + ZnCl2 (20 mol%) | 18 |
| 3 | FeCl3·6H2O/Gly (3:1 mol mol−1) | 23 |
| 4 | ZnCl2/ChCl (1:1 mol mol−1) | 98 c |
| 5 | FeCl3·6H2O/urea (2:1 mol mol−1) | 98 |
| 6 | PTSA/ChCl (2:1 mol mol−1) | 83 |
| Reaction Solvent | Yield (%) | AE (%) | RME (%) | OE (%) | EM (%) | PMIRXN c (g g−1) | PMIWU d (g g−1) | RI e | RP (%) | E-Factor f |
|---|---|---|---|---|---|---|---|---|---|---|
| Cy, EtOAc, MeOH b | 98 | 92 | 15 | 16 | 8.3 | 7.2 | 48.3 | 12.2 | 25.2 | 20 |
| ZnCl2/ChCl g | 98 | 92 | 89 | 97 | 36.3 | 4.2 | 20.0 | 13.3 | 66.5 | 7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cicco, L.; Perna, F.M.; Capriati, V.; Vitale, P. A Sustainable Synthetic Approach to Tacrine and Cholinesterase Inhibitors in Deep Eutectic Solvents under Aerobic Conditions. Molecules 2024, 29, 1399. https://doi.org/10.3390/molecules29061399
Cicco L, Perna FM, Capriati V, Vitale P. A Sustainable Synthetic Approach to Tacrine and Cholinesterase Inhibitors in Deep Eutectic Solvents under Aerobic Conditions. Molecules. 2024; 29(6):1399. https://doi.org/10.3390/molecules29061399
Chicago/Turabian StyleCicco, Luciana, Filippo Maria Perna, Vito Capriati, and Paola Vitale. 2024. "A Sustainable Synthetic Approach to Tacrine and Cholinesterase Inhibitors in Deep Eutectic Solvents under Aerobic Conditions" Molecules 29, no. 6: 1399. https://doi.org/10.3390/molecules29061399
APA StyleCicco, L., Perna, F. M., Capriati, V., & Vitale, P. (2024). A Sustainable Synthetic Approach to Tacrine and Cholinesterase Inhibitors in Deep Eutectic Solvents under Aerobic Conditions. Molecules, 29(6), 1399. https://doi.org/10.3390/molecules29061399