

Computational Binding Study Hints at Ecdysone 20-Mono-Oxygenase as the Hitherto Unknown Target for Ring C-Seco Limonoid-Type Insecticides

Abstract
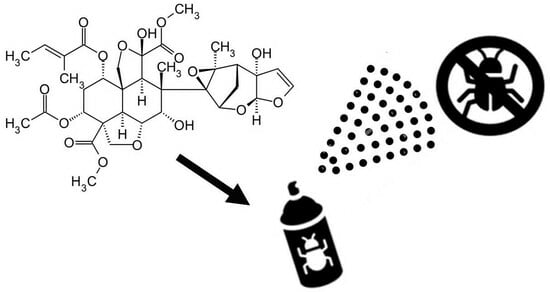
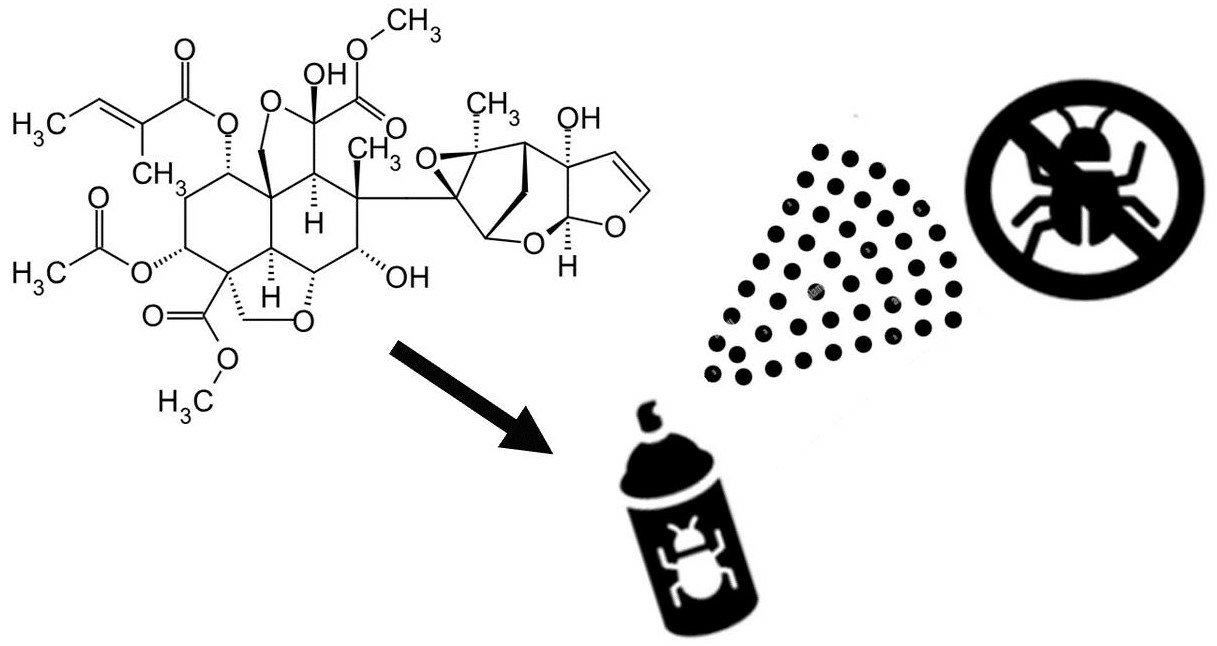
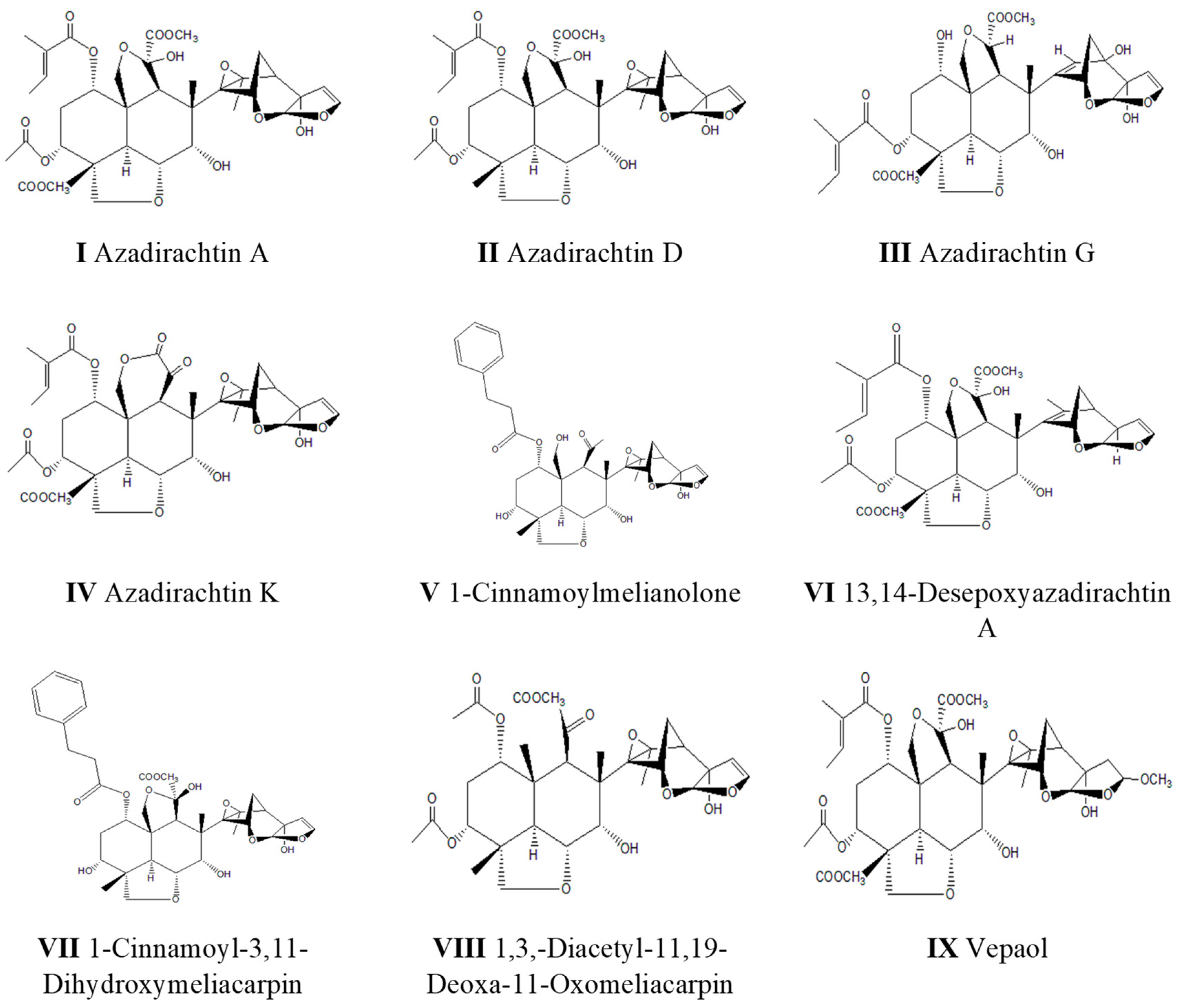
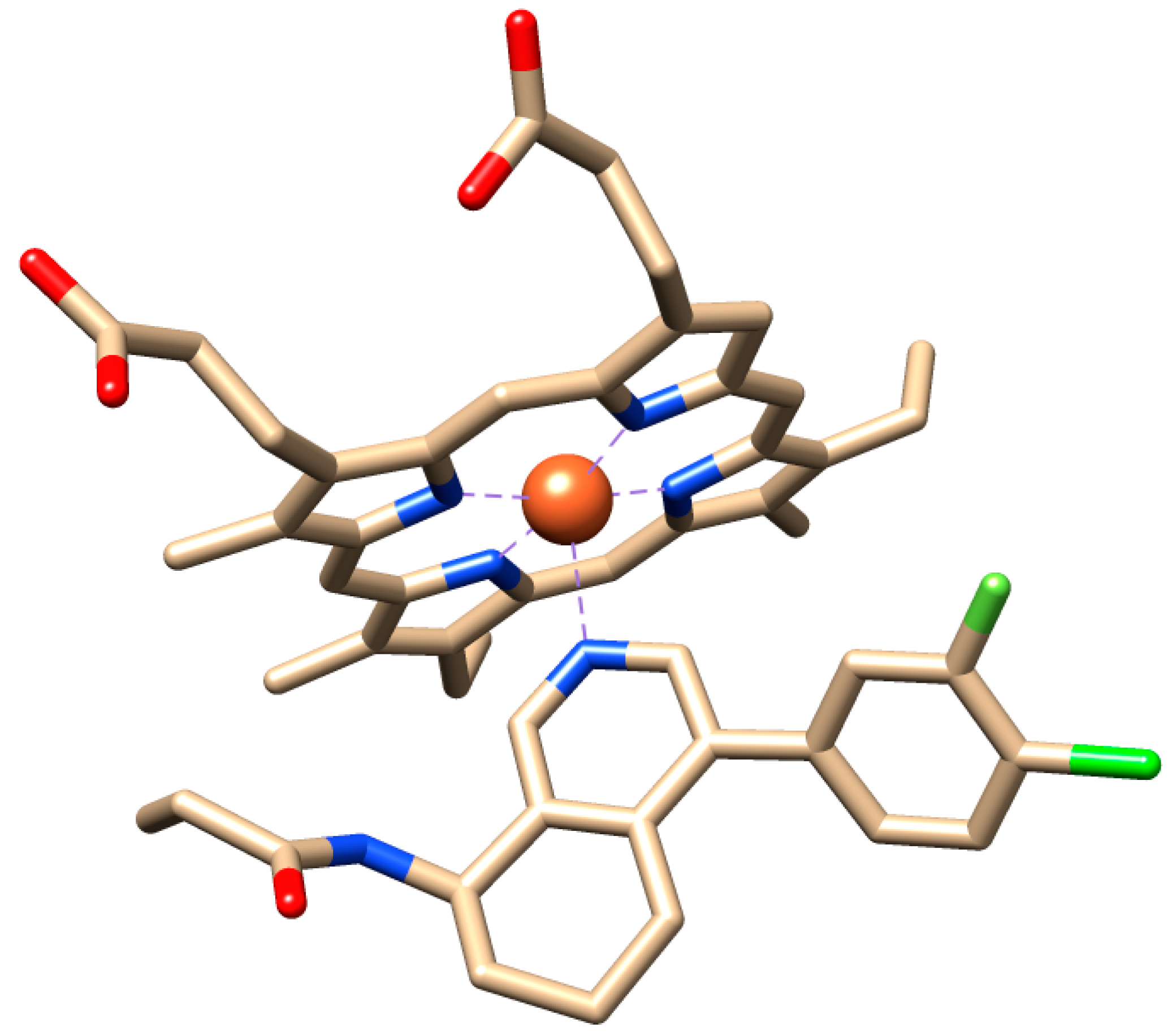
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
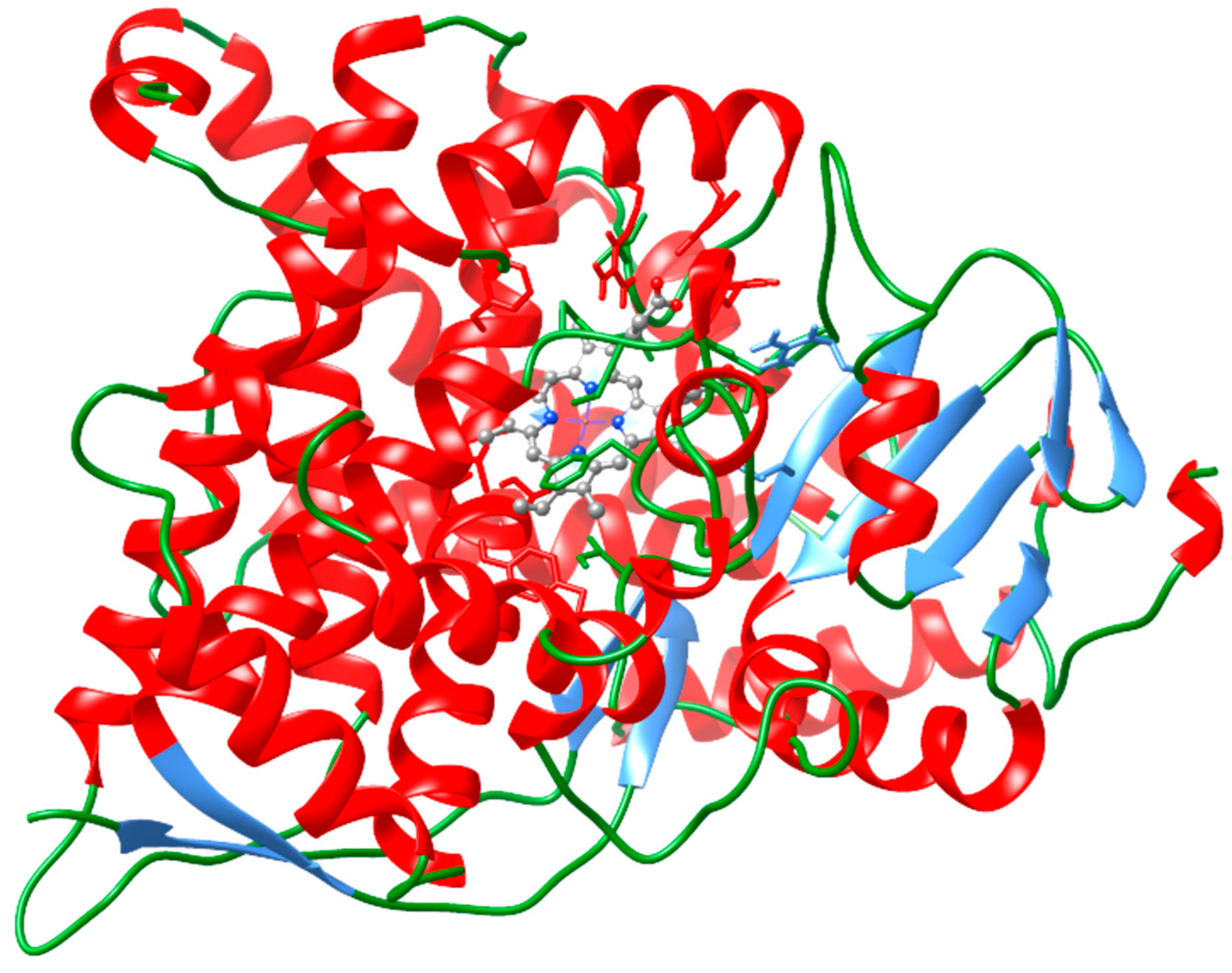
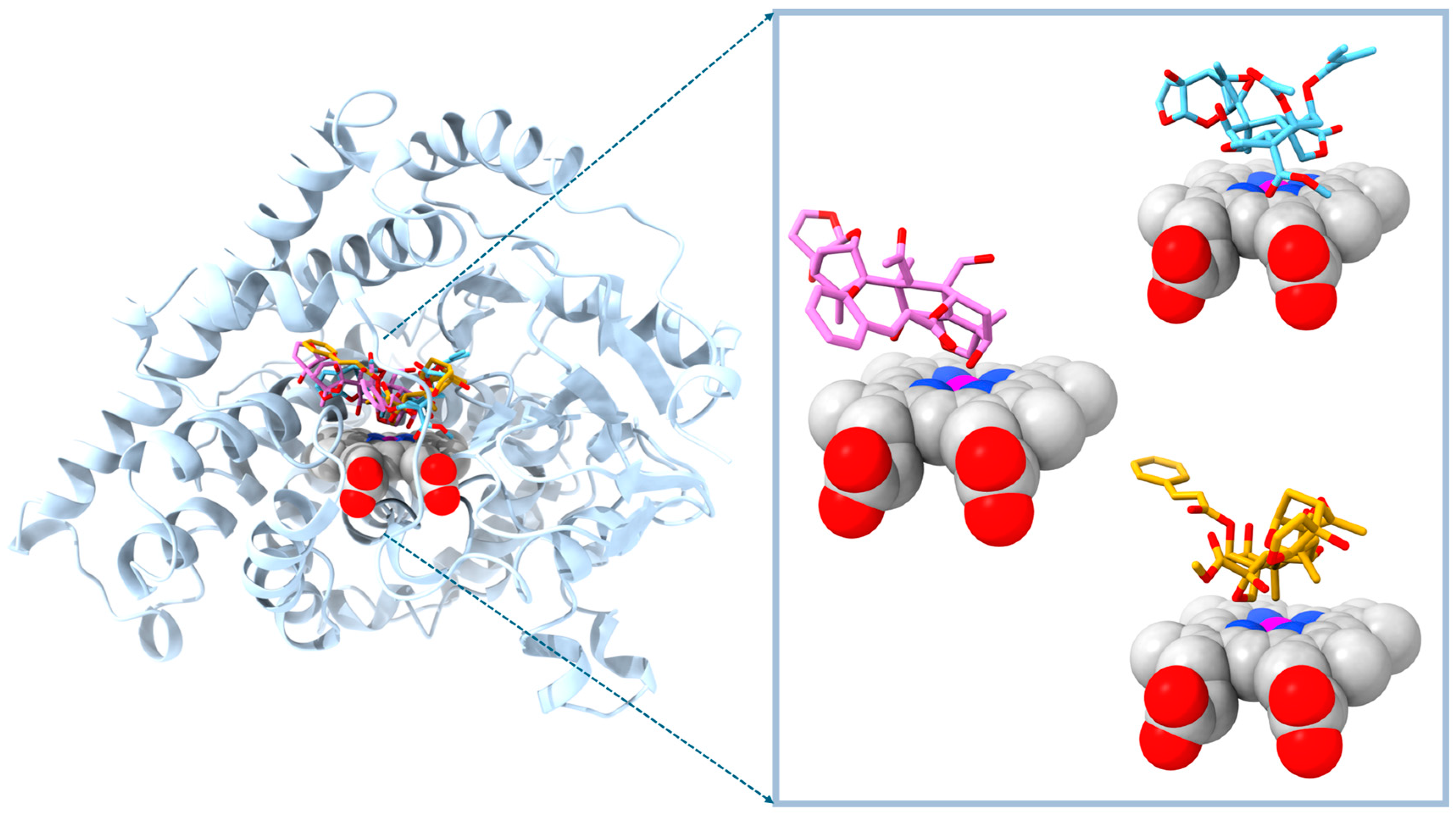
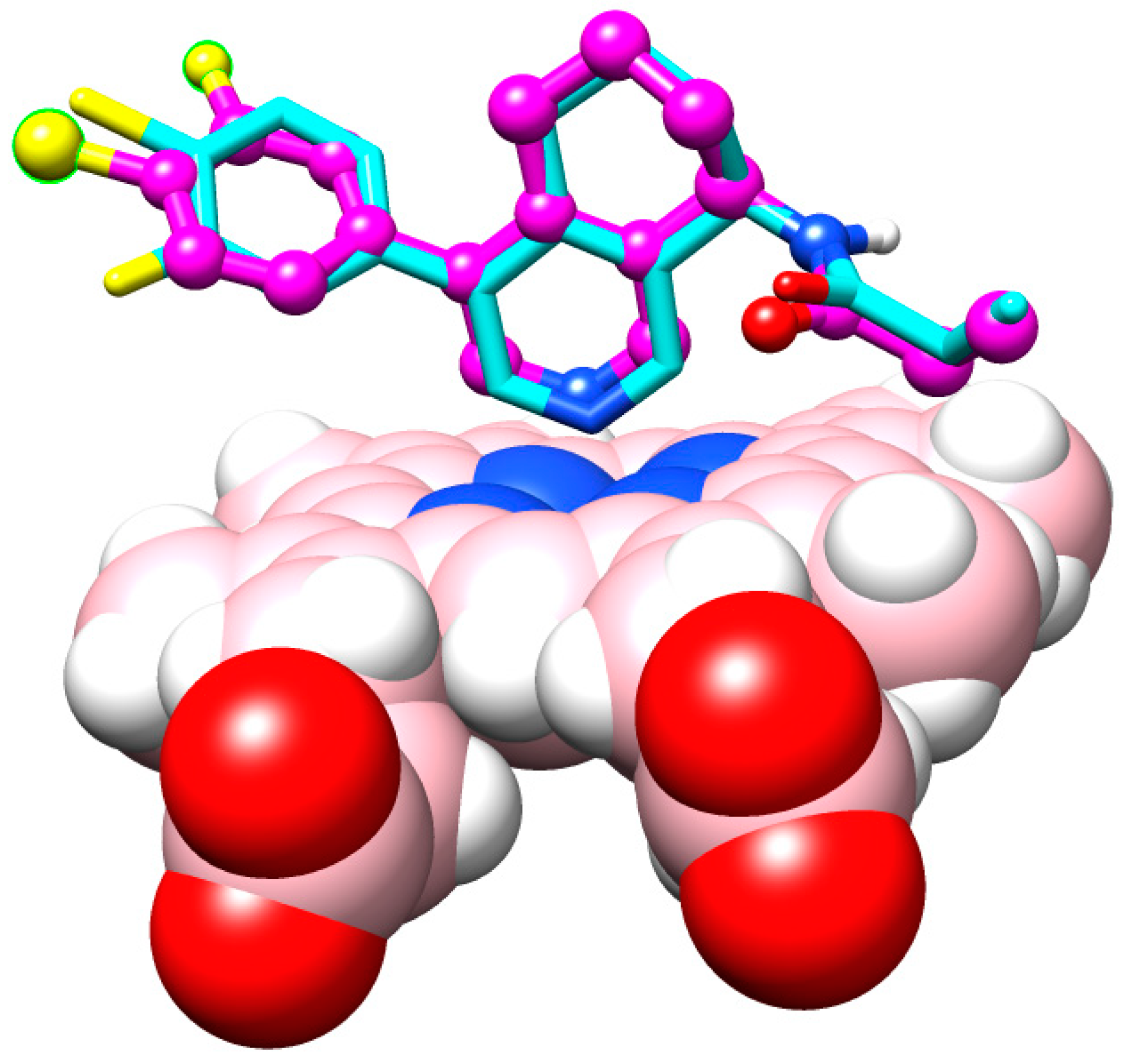
2. Results
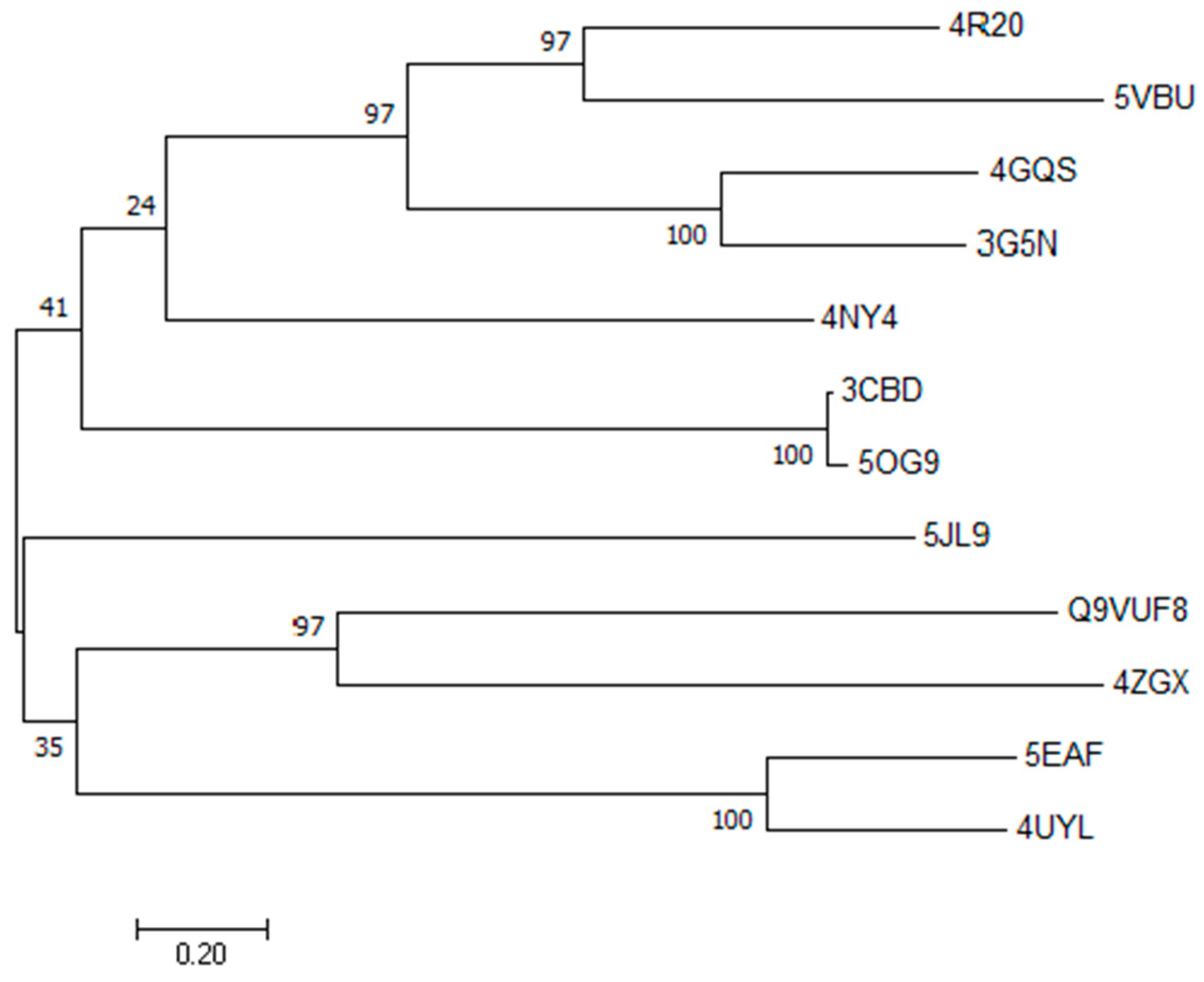
2.1. Target Modeling by Primary Sequence Homology

2.2. Molecular Simulation Details
3. Discussion
4. Materials and Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chandler, D.; Bailey, A.S.; Tatchell, G.M.; Davidson, G.; Greaves, J.; Grant, W.P. The development, regulation and use of biopesticides for integrated pest management. Philos. Trans. R. Soc. B 2011, 366, 1987–1998. [Google Scholar] [CrossRef] [PubMed]
- Drijfhout, F.P.; David Morgan, E. Terrestrial Natural Products as Antifeedants. In Comprehensive Natural Products II; Elsevier: Amsterdam, The Netherlands, 2010; pp. 457–501. [Google Scholar]
- National Research Council (US) Panel on Neem. Neem: A Tree for Solving Global Problems; National Academies Press: Washington, DC, USA, 1992; ISBN 978-0-309-04686-2. [Google Scholar]
- Koul, O. Mode of action of azadirachtin in insects. In NEEM; Randhawa, N.S., Parmar, B.S., Eds.; New Age International: New Delhi, India, 1996; pp. 160–170. [Google Scholar]
- Ley, S.V.; Lovell, H.; Williams, D.J. Chemistry of insect antifeedants from Azadirachta indica, Part 14: Absolute configuration of azadirachtin. J. Chem. Soc. Chem. Commun. 1992, 18, 1304–1306. [Google Scholar] [CrossRef]
- Gualdani, R.; Cavalluzzi, M.M.; Lentini, G.; Habtemariam, S. The Chemistry and Pharmacology of Citrus Limonoids. Molecules 2016, 21, 1530. [Google Scholar] [CrossRef]
- Mordue (Luntz), A.J.; Nisbet, A.J. Azadirachtin from the neem tree Azadirachta indica: Its action against insects. An. Soc. Entomol. Bras. 2000, 29, 615–632. [Google Scholar] [CrossRef]
- Senthil-Nathan, S. Physiological and Biochemical Effect of Neem and Other Meliaceae Plants Secondary Metabolites against Lepidopteran Insects. Front. Physiol. 2013, 4, 359. [Google Scholar] [CrossRef]
- Roy, A.; Saraf, S. Limonoids: Overview of significant bioactive triterpenes distributed in plants kingdom. Biol. Pharm. Bull. 2006, 29, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.D. Azadirachtin, a scientific gold mine. Bioorgan. Med. Chem. 2009, 17, 4096–4105. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.-G.; Luo, X.-D. Meliaceous Limonoids: Chemistry and Biological Activities. Chem. Rev. 2011, 111, 7437–7522. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision, C.01 and GaussView6; Gaussian, Inc.: Wallingford, CT, USA, 2016; Available online: http://gaussian.com (accessed on 14 July 2023).
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- NCBI. BLAST>> Blastp Suite. U.S. National Library of Medicine. Available online: https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins (accessed on 10 November 2023).
- Pallan, P.S.; Nagy, L.D.; Lei, L.; Gonzalez, E.; Kramlinger, V.M.; Azumaya, C.M.; Wawrzak, Z.; Waterman, M.R.; Guengerich, F.P.; Egli, M. Structural and Kinetic Basis of Steroid 17α,20-Lyase Activity in Teleost Fish Cyto-chrome P450 17A1 and Its Absence in Cytochrome P450 17A2. J. Biol. Chem. 2015, 290, 3248–3268. [Google Scholar] [CrossRef]
- Fasan, R. Directed Evolution of Cytochrome P450 BM3, to Octane Monoxygenase 139-3. To be published. Protein Data Bank 2008. Available online: https://www.rcsb.org/structure/3CBD (accessed on 19 November 2023).
- Wang, C.; Pallan, P.S.; Zhang, W.; Lei, L.; Yoshimoto, F.K.; Waterman, M.R.; Egli, M.; Guengerich, F.P. Functional Analysis of Human Cytochrome P450 21A2 Variants Involved in Congenital Adrenal Hyperplasia. J. Biol. Chem. 2017, 292, 10767–10778. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.E.; Aebi, J.D.; Hornsperger, B.; Krebs, H.; Kuhn, B.; Kuglstatter, A.; Alker, A.M.; Peter, H.; Mu, S.; Burger, D.; et al. Discovery of 4 Aryl-5,6,7,8-Tetrahydroisoquinolines as Potent, Selective, and Orally Active Aldosterone Synthase (CYP11B2) Inhibitors: In Vivo Evaluation in Rodents and Cynomolgus Monkeys. J. Med. Chem. 2015, 8054–8065. [Google Scholar] [CrossRef] [PubMed]
- Acevedo-Rocha, C. Regio- and Diastereoselective Hydroxylation of Steroids Using P450-BM3: Readdressing the Numbers Problem in Directed Evolution. To be published. Protein Data Bank 2018. Available online: https://www.rcsb.org/structure/5og9 (accessed on 19 November 2023).
- Tyndall, J.D.A.; Sabherwal, M.; Sagatova, A.A.; Keniya, M.V.; Negroni, J.; Wilson, R.K.; Woods, M.A.; Tietjen, K.; Monk, B.C. Structural and Functional Elucidation of Yeast Lanosterol 14α-Demethylase in Complex with Agro-chemical Antifungals. PLoS ONE 2016, 11, e0167485. [Google Scholar] [CrossRef]
- Ghosh, D.; Egbuta, C.; Lo, J. Testosterone Complex and Non-Steroidal Ligands of Human Aromatase. J. Steroid Biochem. Mol. Biol. 2018, 181, 11–19. [Google Scholar] [CrossRef]
- Hargrove, T.Y.; Wawrzak, Z.; Lamb, D.C.; Guengerich, F.P.; Lepesheva, G.I. Structure-Functional Characterization of Cytochrome P450 Sterol 14α-Demethylase (CYP51B) from Aspergillus Fumigatus and Molecular Basis for the Development of Antifungal Drugs. J. Biol. Chem. 2015, 290, 23916–23934. [Google Scholar] [CrossRef]
- Reynald, R.L.; Sansen, S.; Stout, C.D.; Johnson, E.F. Structural Characterization of Human Cytochrome P450 2C19. J. Biol. Chem. 2012, 287, 44581–44591. [Google Scholar] [CrossRef] [PubMed]
- Gay, S.C.; Sun, L.; Maekawa, K.; Halpert, J.R.; Stout, C.D. Crystal Structures of Cytochrome P450 2B4 in Complex with the Inhibitor 1-Biphenyl-4-Methyl-1 H -Imidazole: Ligand-Induced Structural Response through α-Helical Repositioning. Biochemistry 2009, 48, 4762–4771. [Google Scholar] [CrossRef]
- Brändén, G.; Sjögren, T.; Schnecke, V.; Xue, Y. Structure-Based Ligand Design to Overcome CYP Inhibition in Drug Discovery Projects. Drug Discov. Today 2014, 19, 905–911. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Henderson, A.R. The Bootstrap: A Technique for Data-Driven Statistics. Using Computer-Intensive Analyses to Explore Experimental Data. Clin. Chim. Acta 2005, 359, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The Rapid Generation of Mutation Data Matrices from Protein Sequences. Bioinformatics 1992, 8, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology Modelling of Protein Structures and Complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An Environment for Comparative Protein Modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Quiroga, I.; Scior, T. Structure–Function Analysis of the Cytochromes P450, Responsible for Phenprocoumon Metabolism. J. Mex. Chem. Soc. 2018, 61, 349–360. [Google Scholar] [CrossRef]
- Quiroga, I.; Kammerer, F.M.; Scior, T. Identification of a New Site of Metabolism for Phenprocoumon by Modeling Its CYP2C9 Hydroxylation Pattern. SAJ Pharm. Pharmacol. 2018, 5, 1–2. [Google Scholar]
- Scior, T.; Quiroga, I.; Kammerer, B. Inquiry of Literature Evidence for Induced Fit and Regioselectivity of Cyto-chrome P450 Enzyme CYP2C9: A Critical Review. SCIOL Biotechnol. 2018, 1, 30–48. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- DeMars, M.D.; Sheng, F.; Park, S.R.; Lowell, A.N.; Podust, L.M.; Sherman, D.H. Biochemical and Structural Char-acterization of MycCI, a Versatile P450 Biocatalyst from the Mycinamicin Biosynthetic Pathway. ACS Chem. Biol. 2016, 11, 2642–2654. [Google Scholar] [CrossRef]
- Strushkevich, N. Crystal Structure of Human CYP7A1 in Complex with Cholest-4-En-3-One. Protein Data Bank 2011. Available online: https://www.ncbi.nlm.nih.gov/Structure/pdb/3SN5 (accessed on 19 November 2023).
- Schlichting, I.; Berendzen, J.; Chu, K.; Stock, A.M.; Maves, S.A.; Benson, D.E.; Sweet, R.M.; Ringe, D.; Petsko, G.A.; Sligar, S.G. The Catalytic Pathway of Cytochrome P450cam at Atomic Resolution. Science 2000, 287, 1615–1622. [Google Scholar] [CrossRef]
- Meharenna, Y.T.; Li, H.; Hawkes, D.B.; Pearson, A.G.; De Voss, J.; Poulos, T.L. Crystal Structure of P450cin in a Complex with Its Substrate, 1,8-Cineole, a Close Structural Homologue to d-Camphor, the Substrate for P450cam. Biochemistry 2004, 43, 9487–9494. [Google Scholar] [CrossRef] [PubMed]
- Strushkevich, N.; Gilep, A.A.; Shen, L.; Arrowsmith, C.H.; Edwards, A.M.; Usanov, S.A.; Park, H.-W. Structural Insights into Aldosterone Synthase Substrate Specificity and Targeted Inhibition. Mol. Endocrinol. 2013, 27, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Stout, C.D.; Zhang, Q.; Johnson, E.F. Contributions of Ionic Interactions and Protein Dynamics to Cytochrome P450 2D6 (CYP2D6) Substrate and Inhibitor Binding. J. Biol. Chem. 2015, 290, 5092–5104. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.-J.; Kim, K.; Kim, C.; Kim, M.-J.; Kim, T.-O.; Lee, S.-E. Molecular Mechanisms of Anti-Melanogenic Ge-dunin Derived from Neem Tree (Azadirachta indica) Using B16F10 Mouse Melanoma Cells and Early-Stage Zebrafish. Plants 2021, 10, 330. [Google Scholar] [CrossRef] [PubMed]
- Milani, G.; Cavalluzzi, M.M.; Solidoro, R.; Salvagno, L.; Quintieri, L.; Di Somma, A.; Rosato, A.; Corbo, F.; Franchini, C.; Duilio, A.; et al. Molecular Simplification of Natural Products: Synthesis, Antibacterial Activity, and Molecular Docking Studies of Berberine Open Models. Biomedicines 2021, 9, 452. [Google Scholar] [CrossRef] [PubMed]
- Tundis, R.; Xiao, J.; Silva, A.S.; Carreiró, F.; Loizzo, M.R. Health-Promoting Properties and Potential Application in the Food Industry of Citrus medica L. and Citrus × clementina Hort. Ex Tan. Essential Oils and Their Main Constituents. Plants 2023, 12, 991. [Google Scholar] [CrossRef]
- Jaoko, V.; Nji Tizi Taning, C.; Backx, S.; Mulatya, J.; Van den Abeele, J.; Magomere, T.; Olubayo, F.; Mangelinckx, S.; Werbrouck, S.P.O.; Smagghe, G. The Phytochemical Composition of Melia volkensii and Its Potential for Insect Pest Management. Plants 2020, 9, 143. [Google Scholar] [CrossRef]
- Bankova, V.; Popova, M. Propolis: Harnessing Nature’s Hidden Treasure for Sustainable Agriculture. Agrochemicals 2023, 2, 581–597. [Google Scholar] [CrossRef]
- Ambrus, Á.; Vásárhelyi, A.; Ripka, G.; Szemánné-Dobrik, H.; Szenczi-Cseh, J. Evaluation of the Results of Pesticide Residue Analysis in Food Sampled between 2017 and 2021. Agrochemicals 2023, 2, 409–435. [Google Scholar] [CrossRef]
- Ambrus, Á.; Szenczi-Cseh, J.; Doan, V.V.N.; Vásárhelyi, A. Evaluation of Monitoring Data in Foods. Agrochemicals 2023, 2, 69–95. [Google Scholar] [CrossRef]
- ChemSpider. Available online: http://www.chemspider.com/ (accessed on 11 November 2023).
- Hartree, D.R. The Wave Mechanics of an Atom with a Non-Coulomb Central Field. Part I. Theory and Methods. Math. Proc. Camb. Philos. Soc. 1928, 24, 89–110. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Leach, A.R. Molecular Modelling Principles and Applications, 2nd ed.; Prentice Hall: Hoboken, NJ, USA, 2001; ISBN 0-582-38210-6. [Google Scholar]
- Scior, T.; Wahab, A. Structure Prediction of Proteins with Very Low Homology: A Comprehensive Introduction and a Case Study on Aminopeptidase. Drug Des. Res. Perspect. 2007, 26, 675–708. [Google Scholar]
- Scior, T.; Verhoff, M.; Gutierrez-Aztatzi, I.; Ammon, H.P.T.; Laufer, S.; Werz, O. Interference of Boswellic Acids with the Ligand Binding Domain of the Glucocorticoid Receptor. J. Chem. Inf. Model. 2014, 54, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Bumbăcilă, B.; Putz, M.V. Neurotoxicity of Pesticides: The Roadmap for the Cubic Mode of Action. Curr. Med. Chem. 2020, 27, 54–77. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramírez, R.E.; Buendia-Corona, R.E.; Pérez-Xochipa, I.; Scior, T. Computational Binding Study Hints at Ecdysone 20-Mono-Oxygenase as the Hitherto Unknown Target for Ring C-Seco Limonoid-Type Insecticides. Molecules 2024, 29, 1628. https://doi.org/10.3390/molecules29071628
Ramírez RE, Buendia-Corona RE, Pérez-Xochipa I, Scior T. Computational Binding Study Hints at Ecdysone 20-Mono-Oxygenase as the Hitherto Unknown Target for Ring C-Seco Limonoid-Type Insecticides. Molecules. 2024; 29(7):1628. https://doi.org/10.3390/molecules29071628
Chicago/Turabian StyleRamírez, Ramsés E., Ricardo E. Buendia-Corona, Ivonne Pérez-Xochipa, and Thomas Scior. 2024. "Computational Binding Study Hints at Ecdysone 20-Mono-Oxygenase as the Hitherto Unknown Target for Ring C-Seco Limonoid-Type Insecticides" Molecules 29, no. 7: 1628. https://doi.org/10.3390/molecules29071628
APA StyleRamírez, R. E., Buendia-Corona, R. E., Pérez-Xochipa, I., & Scior, T. (2024). Computational Binding Study Hints at Ecdysone 20-Mono-Oxygenase as the Hitherto Unknown Target for Ring C-Seco Limonoid-Type Insecticides. Molecules, 29(7), 1628. https://doi.org/10.3390/molecules29071628